
伴骨骼肌损害的嗜酸性肌筋膜炎5例临床病理分析
2022-01-19 08:31瞿千千王孟丽刘海燕吕海东
临床与实验病理学杂志 2021年12期
范 云,瞿千千,王孟丽,刘海燕,吕海东
嗜酸性肌筋膜炎(eosinophilic fasciitis, EF)于1974年由Shulman首次提出,是以肌筋膜弥漫性肿胀和硬化,常伴嗜酸细胞增高为特征的结缔组织病,属炎性肌病的一种特殊类型。国内自1981年以来有个别病例报道[1-2],但合并骨骼肌损害的EF文献报道较少见。本文着重探讨伴骨骼肌损害EF患者的临床和病理资料,并结合相关文献进行讨论,旨在提高临床和病理医师对EF的认识水平,减少误诊、误治的发生。
1 材料与方法
1.1 临床资料收集2006年1月~2019年12月焦作市人民医院神经内科门诊和住院治疗的伴骨骼肌损害的EF 5例,均为男性。亚急性起病2例,慢性起病逐渐进展3例。病程1个月~1.5年,平均5.3个月。
1.2 方法
1.2.1免疫组化 采用免疫组化EnVision法检测淋巴细胞CD4、CD8、CD20和CD68的表达,镜下观察。
1.2.2神经电生理 采用日本光电MEB-9200K肌电诱发电位仪,常规行同心圆针电极肌电图和感觉运动神经传导速度检查。
1.2.3肌肉病理 在患者家属签署知情同意书后,局麻下开放性活检取肌肉和肌筋膜组织,大小0.5 cm×0.5 cm×1.0 cm。活检部位:肱二头肌2例,腓肠肌2例,股四头肌1例。肌肉标本经液氮固定冷冻切片,行常规组织学和酶组织化学染色。染色包括HE、改良高墨瑞(MGT)、PAS、油红“O”、ATP酶(酸性pH 4.3,碱性pH 10.6)、还原型辅酶Ⅰ(NADH-TR)等染色。并采用免疫组化EnVision法检测淋巴细胞CD4、CD8、CD20和CD68的表达。
2 结果
2.1 临床特点5例患者临床表现为不同程度的肢体肿胀、肌肉疼痛和肢体无力,且均以肢体远端为重。其中3例患者双上肢远端肌力3~4级,双手握拳困难,呈“空心拳”征。2例患者出现双下肢远端无力,双足背伸肌力3级。伴肌肉疼痛4例,关节疼痛2例(表1)。4例患者可见肢体局部皮肤有色素沉着,呈类似橘皮样改变,色素沉着呈斑片状分布(图1)。
2.2 实验室检查3例患者外周血嗜酸性粒细胞升高,3例血清肌酸激酶升高(956~1 711 U/L)。C反应蛋白(CRP)、血沉(ESR)升高各2例(表1)。
2.3 肌电图5例患者中4例肌电图呈肌源性损害,1例未见神经源性及肌源性损害。被检测的感觉神经和运动神经传导速度均正常。
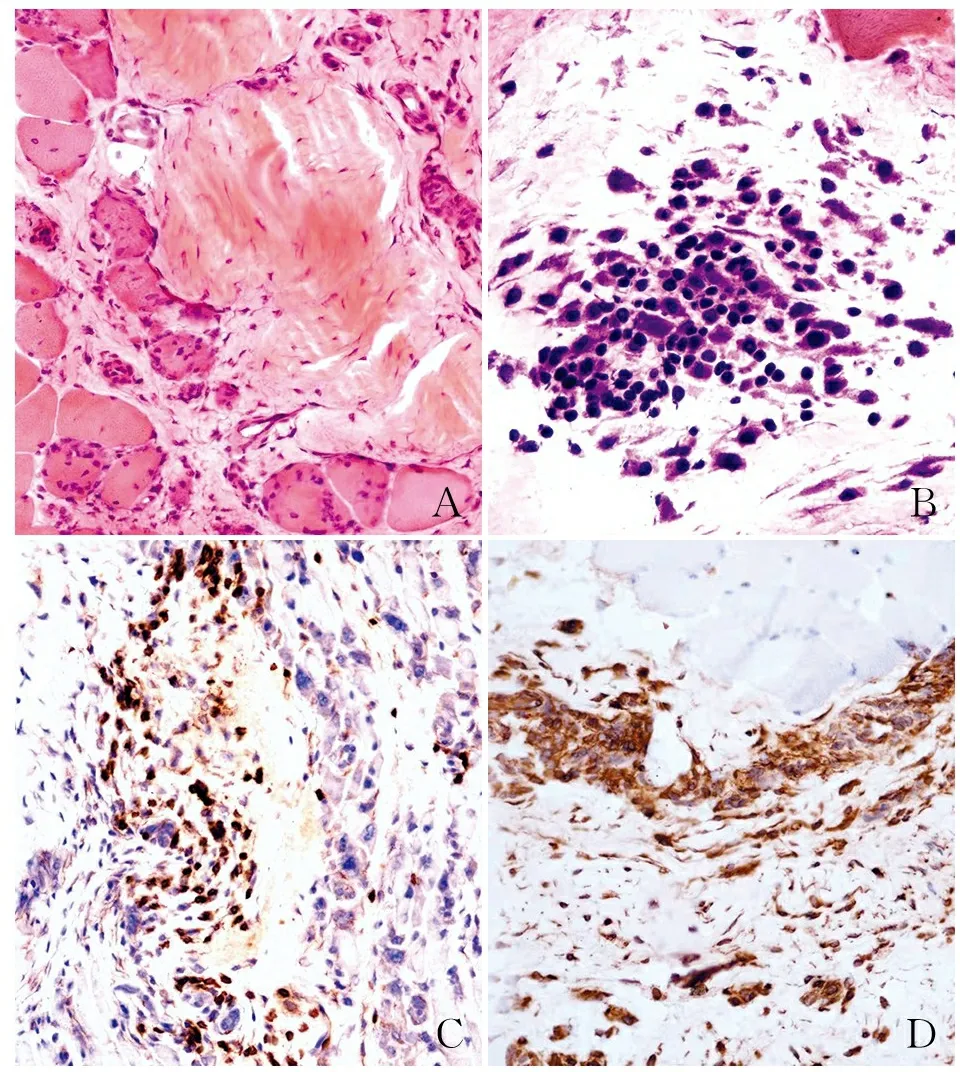
2.4 组织学特征5例患者肌筋膜均有不同程度增厚,伴小血管轻度增多,4例与深筋膜相邻的肌纤维有变性坏死,伴炎性细胞浸润(图2A),2例可见嗜酸性粒细胞在肌筋膜组织中聚集(图2B)。淋巴细胞亚群免疫组化染色可见肌筋膜和肌组织中的炎性细胞主要为CD8、CD68+淋巴细胞(图2C、D),CD20+淋巴细胞少见。5例患者肌肉组织ATP酶染色提示Ⅰ、Ⅱ型肌纤维分布正常,MGT、油红“O”、NADH-TR、PAS染色均未见特殊改变。
ABCD
3 讨论
EF是由于肌筋膜软组织出现无菌性炎症而引起的慢性免疫性疾病[3],可发生于任何年龄,男性明显多于女性[4-5]。本组5例患者均为男性,除1例59岁患者外,其余4例年龄为17~26岁,提示EF好发于青年男性。EF一般为亚急性或慢性起病[4],部分患者在发病前有劳累受凉病史[4,6]。EF的主要临床表现为四肢皮肤和皮下组织肿胀疼痛,病变常累及前臂、上臂、小腿和大腿等部位,而手足和面部通常不受累[7-9]。部分患者可有皮肤色素沉着,呈类似橘皮样改变[10]。本组5例患者中有4例出现皮肤及皮下组织损害,表现为皮肤肿胀发硬,3例皮肤有斑片状色素沉着,部分呈橘皮样改变,亦说明皮肤损害是EF常见的临床体征,橘皮样改变具有一定的特异性。
伴有骨骼肌损害的EF患者除皮肤损害以外,常伴有不同程度的肢体无力。本组5例患者中3例出现双上肢远端无力,双手握拳困难;2例出现双下肢远端无力,双足背伸无力,提示EF患者合并有肌肉损害;4例患者有不同程度的肌肉疼痛,肌电图呈肌源性损害,3例血清肌酸激酶明显升高,均支持EF患者有骨骼肌组织病变。EF患者周围血象中部分可见嗜酸性粒细胞升高,但并非每例患者均升高,本组5例患者中仅3例外周血嗜酸性粒细胞升高。有文献报道嗜酸性粒细胞升高可能与患者病程长短有一定关系[11]。
EF的组织病理改变主要在深筋膜,其特征性病理改变为肌筋膜组织水肿、增厚和炎性细胞浸润[12-13]。本组5例EF均可见肌筋膜有不同程度的小血管增多和炎性细胞浸润,有2例可见嗜酸性粒细胞在筋膜组织中聚集现象。与以往文献报道不同的是,本组有4例患者在与肌筋膜相邻的骨骼肌组织中出现部分肌纤维变性坏死,并伴有炎性细胞浸润,呈明显肌肉损害的病理改变,作者认为这是造成患者肌酸激酶升高和肢体无力的主要原因。本组1例患者临床上有肌无力症状,血清肌酸激酶明显升高,电生理检查提示肌源性损害,但组织活检中仅见部分肌筋膜有炎性细胞浸润,而未见肌纤维变性坏死,作者认为这与肌纤维坏死呈灶性分布和组织活检部位的选择有一定关系。
淋巴细胞亚群免疫组化染色可见肌筋膜内和小血管周围的炎性细胞主要为CD8+和CD68+淋巴细胞,而CD20+淋巴细胞少见,提示EF可能是以细胞毒性免疫反应为主。有文献报道[8]肌筋膜炎早期的病理改变较轻,可见肌筋膜内有炎性细胞浸润和小血管增多,以及嗜酸性粒细胞浸润。随着疾病的发展可出现肌纤维变性坏死和硬化萎缩,此时嗜酸性粒细胞常消失,本组仅有2例查见肌筋膜组织内有嗜酸性粒细胞聚集特征性病理改变。虽然本组未对同一患者的不同病程阶段多次取材来观察组织中的炎性病理变化,但可以注意到病程较长患者的肌纤维变性坏死的程度较为明显。因此,对EF患者早期发现、早期诊断具有重要的临床意义,能够及时应用免疫抑制剂治疗,有效避免相邻骨骼肌组织的进一步损害。
EF是一种以皮肤、筋膜和肌肉损害为主的非特异性炎性疾病,临床上需要与皮肌炎、嗜酸细胞性多发性肌炎、硬皮病等疾病相鉴别[8,14]。EF的治疗以免疫抑制剂治疗为主,本组5例患者均应用糖皮质激素治疗,激素治疗2~4周后,患者临床症状开始减轻,运动功能逐渐改善;维持治疗6~8周后,激素剂量开始逐渐减量直至停用。
综上所述,EF不仅有皮肤和皮下组织病变,还可出现相邻骨骼肌的损害。对EF患者进行早期诊断和治疗,可有效避免或减轻骨骼肌组织的损害,肌肉和筋膜组织活检是早期诊断EF的重要手段。
猜你喜欢
中国耳鼻咽喉颅底外科杂志(2022年4期)2022-11-19
体育科技文献通报(2022年3期)2022-05-23
医学综述(2021年16期)2021-12-01
国际放射医学核医学杂志(2021年10期)2021-02-28
家庭百事通·健康一点通(2020年10期)2020-10-27
恋爱婚姻家庭·养生版(2020年9期)2020-10-20
家庭医药·快乐养生(2020年9期)2020-09-27
灌篮(2020年36期)2020-05-16
运动(2018年14期)2018-07-16
体育科学(2018年3期)2018-04-20
