
原发性骨弥漫大B细胞淋巴瘤15例临床病理特征及预后
2022-01-19 08:31梁小芹苏勤军冯友繁王慧春王金穗
临床与实验病理学杂志 2021年12期
梁小芹,苏勤军,冯友繁,王慧春,王 卓,王金穗
原发性骨淋巴瘤(primary bone lymphoma, PBL)是一种非常少见的结外淋巴瘤,占恶性骨肿瘤的7%,占结外淋巴瘤的5%[1]。PBL临床表现缺乏特异性,常见症状为疼痛、局部肿块及骨折[2]。WHO(2020)骨与软组织分类[3]将PBL定义为恶性淋巴细胞构成的骨内肿瘤性病变,病变累及1处或多处骨骼部位;无区域淋巴结或结外病变受累的证据。PBL最常见病理类型是弥漫大B细胞淋巴瘤(diffuse large B-cell lymphoma, DLBCL),占骨原发性淋巴瘤的95%[4]。原发性骨弥漫大B细胞淋巴瘤(primary bone diffuse large B-cell lymphoma, PB-DLBCL)发病率低,临床表现缺乏特异性,目前尚无标准的治疗规范。因此,本文收集15例PB-DLBCL,对其临床表现、诊断、治疗和转归进行回顾性分析,以提高病理及临床医师对该病的认识。
1 材料与方法
1.1 临床资料收集2013年1月~2020年12月甘肃省人民医院收治的15例PB-DLBCL,患者临床资料均完整。PB-DLBCL的诊断和分类采用WHO(2020)造血与淋巴组织肿瘤分类标准,临床分期依据Ann Arbor分期标准。
1.2 方法
1.2.1免疫组化 对石蜡包埋组织切片行常规HE染色,显微镜下观察。免疫组化染色采用EnVision两步法,一抗包括CKpan、EMA、vimentin、CD3、CD5、CD20、CD79a、MUM1、CD10、BCL-2、BCL-6、CD138、PAX-5、c-myc、Ki-67,均购自福州迈新公司。肿瘤细胞核/质(膜)出现清晰棕黄色颗粒为阳性细胞,其中BCL-2阳性细胞≥50%为阳性,CD10、BCL-6、MUM1阳性细胞≥30%为阳性,c-myc阳性细胞≥40%为阳性,实验均设阴、阳性对照。
1.2.2EBER原位杂交 EBER原位杂交试剂盒购自罗氏公司,操作按试剂盒说明书步骤进行,分别选用鼻咽部NK/T细胞淋巴瘤组织和增生扁桃体的淋巴组织作为阳性和阴性对照。
1.2.3FISH BCL-2、BCL-6及c-myc基因断裂双色探针和双色融合探针检测试剂盒及样本释放剂均购自武汉康录生物公司,操作步骤及结果判断按试剂盒说明书进行。正常情况下,1个细胞核内显示2个黄色融合信号;发生重排时,1个细胞核内显示1个黄色融合信号,并可见1个红色和1个绿色分离信号,出现分离信号的肿瘤细胞数≥15%判定为阳性。以有BCL-2、BCL-6和c-myc多拷贝及断裂的样本作为阳性对照,以正常淋巴结为阴性对照。
1.3 疗效评价按照非霍奇金淋巴瘤Lugano疗效评价标准评价,包括完全缓解(complete response, CR)、部分缓解(partial response, PR)、稳定(stable disease, SD)和进展(progressive disease, PD)。CR+PR所占比率为总有效率(overall response, OR)。无进展生存期(progressive-free susvial, PFS)为患者确诊至疾病进展、死亡或随访结束的时间。总生存期(overal susvial, OS)为患者确诊至患者死亡或随访终点的时间。
1.4 随访所有患者通过门诊或电话形式进行随访,随访截至2020年12月31日,失访病例以失访时间为随访终点。
2 结果
2.1 临床特点15例PB-DLBCL患者年龄19~89岁,中位年龄57岁。其中男性7例,女性8例。病灶位于颌骨6例;扁骨5例,包括髂骨2例,肋骨1例,椎骨1例,跖骨1例;长骨者4例,包括肱骨1例,股骨2例,胫骨1例。13例为单发病灶,2例为多发病灶。临床表现为局部疼痛9例,局部包块者3例,3例活动受限,3例伴有B症状。X线和CT表现为不同程度的溶骨性破坏;MRI显示T1W1不规则斑点状或片状低信号,T2W1呈高信号。4例患者行PET-CT检查显示病变区骨质破坏,FDG代谢不均匀性异常增高。Ann Arbor临床分期:Ⅰ期8例,Ⅱ期2例,Ⅳ期5例。国际预后指数(IPI)评分1分者2例,2分者5例,3分者4例,4分者3例,5分者1例(表1)。
2.2 镜检镜下见正常骨结构消失,骨皮质破坏,大的淋巴细胞样肿瘤细胞弥漫分布于骨小梁之间(图1),瘤细胞形态较单一,胞质中等、弱嗜碱或嗜双色性,核大、圆形或椭圆形,核仁明显,有单个居中的核仁或2~4个靠近核膜的小核仁(图2),呈免疫母细胞样或中心母细胞样,核分裂象易见。部分病例伴不规则凝固性坏死,个别病例瘤细胞形态多样。
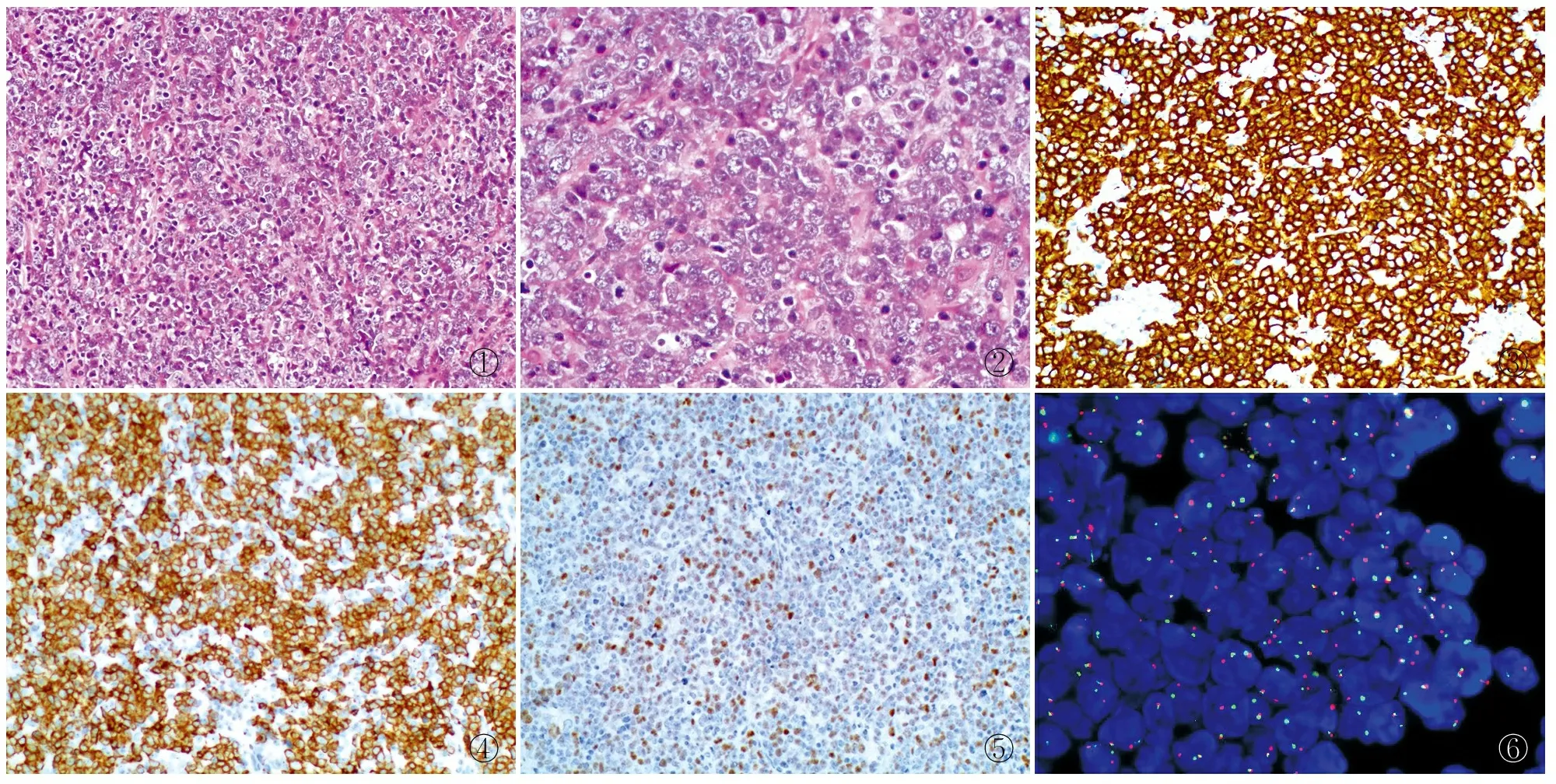
①②③④⑤⑥
2.3 免疫表型肿瘤细胞B细胞标记CD20(图3)、CD79a和PAX-5均阳性,上皮类标记CK、EMA、间叶性标记vimentin及T细胞标记CD3、CD5均阴性。15例PB-DLBCL中CD10阳性6例(40%),BCL-2阳性7例(46.7%,图4),BCL-6阳性11例(73.3%),MUM1阳性7例(46.6%),c-myc阳性6例(46.6%,图5)。免疫分型依照Hans分类:6例(40%)为生发中心(germinal centre B-cell, GCB)来源,9例(60%)为非生发中心(non-germinal centre B-cell, non-GCB)来源。Ki-67增殖指数≥80%者7例(46.6%)。
2.4 原位杂交15例患者EBER原位杂交检测均为阴性。
2.5 FISH检测15例PB-DLBCL行c-myc、BCL-2和BCL-6基因检测,其中2例因蜡块存放时间过长,未能获取可靠的荧光信号。13例中有2例c-myc基因断裂重排(图6),1例BCL-2基因断裂重排,3例BCL-6基因断裂重排。
2.6 治疗与预后15例患者中,1例穿刺活检后未行后续治疗,3例行单纯手术治疗,11例行CHOP及类似化疗方案,其中手术联合化疗7例,单纯化疗4例。11例行CHOP及类似化疗方案,经初始治疗后6例达CR,4例达PR,1例SD,初始治疗后OR为66.7%(10/15)。其中8例接受R-CHOP方案化疗,初始治疗后6例获CR,2例PR,OR为100%,其中1例多灶性病变接受R-CHOP治疗6个周期后,病灶完全消失(图7)。15例患者中位随访时间19(1~38)个月,其中10例存活,3例因疾病进展死亡,1例因感染死亡,1例失访(表1)。
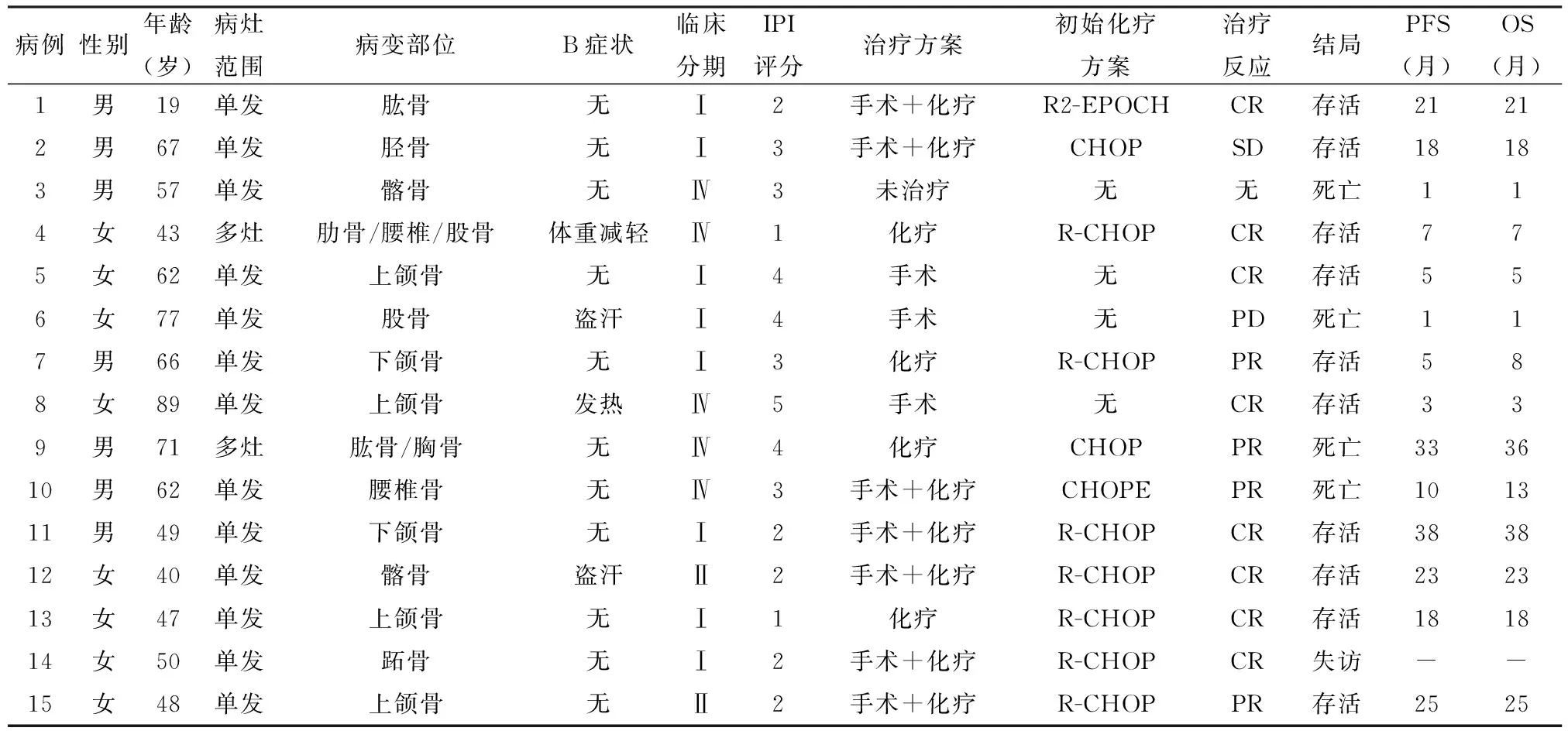
表1 15例原发性骨弥漫大B细胞淋巴瘤的临床特征、治疗及预后

ABCD
3 讨论
PBL于1928年由Oberling首次描述,1971年Shoji等[5]将其命名为PBL并沿用至今。其定义为仅限于骨骼或周围软组织浸润而无其他骨外病变的淋巴瘤。诊断标准:(1)肿瘤发生部位必须是骨骼;(2)临床辅助检查如影像学检查未发现骨骼以外的其他部位淋巴瘤;(3)明确诊断6个月仍未发现骨骼以外的淋巴瘤病灶;(4)必须有明确的病理组织学和免疫组化诊断结果;(5)排除继发性骨淋巴瘤[6]。PBL最常见的病理类型为DLBCL。
目前,PB-DLBCL的病因及发病机制尚不明确,文献报道慢性感染特别是慢性骨髓炎是该病的易感因素[7-8]。PB-DLBCL男性多发,男女比为1.6 ∶1;其可发生于任何年龄,以45~60岁常见,全身骨骼均可受累,以长骨、脊柱和骨盆常见,最常见的临床症状为局部疼痛,其次为局部肿块[9-10]。与全身系统性淋巴瘤相比,PB-DLBCL多为局部表现,全身症状较轻,较少出现B症状。本组15例PB-DLBCL确诊时Ann Arbor临床分期Ⅰ期8例,Ⅱ期2例,Ⅳ期5例,大部分患者分期早,一般情况尚可,考虑可能与患者早期即较为重视有关。
影像学检查是PB-DLBCL的重要辅助检查手段,X线和CT示有不同程度的溶骨性破坏,也可出现成骨性破坏,或两种特征共存[11]。MRI表现为T1W1均一性等或稍低信号,增强后强化[12]。PET-CT可以同时提供解剖和功能学显像,对于观察病变的消退更具优势[13]。但影像学检查缺乏特异性,与其他骨肿瘤、结核及转移癌鉴别有局限性,诊断仍依赖病理组织学检查。
在病理组织学上,PB-DLBC肿瘤细胞浸润性生长,弥漫分布于骨小梁间,瘤细胞大,核卵圆形、卵圆形,可见有单个居中的大核仁或2~4个核仁,核分裂易见,胞质中等量,嗜双色或嗜碱性,免疫组化表达B细胞标记。根据Hans分类标准将DLBCL分为GCB或non-GCB来源,国外研究显示PB-DLBCL中以GCB多见,为non-GCB的1~2倍[2]。本组GCB 6例,non-GCB 9例,与国外报道不一致,但与国内文献报道相近[14]。近年DLBCL的重大进展就是对伴c-myc异常DLBCL的认识。WHO(2016)淋巴瘤分类修订中将c-myc及BCL-2和(或)BCL-6重排的高级别B细胞淋巴瘤作为独立亚型,即双打击或三打击淋巴瘤[15]。有报道发生c-myc基因重排同时伴有BCL-2和(或)BCL-6重排的DLBCL预后差,且对标准化疗(R-CHOP)治疗反应差[16],故分析c-myc基因重排伴BCL-2、BCL-6重排的病例对鉴别预后差的高风险患者至关重要。
PB-DLBCL治疗策略目前尚无统一的标准。CHOP方案及类似方案为目前主要使用的化疗方案,对于CD20+PB-DLBCL患者联合应用利妥昔单抗治疗(即R-CHOP方案),其8年PFS和OS率分别可达100%和95%[17]。本组8例患者接受R-CHOP方案化疗,初始治疗后6例获CR,2例PR,OR为100%,其中最长无进展生存时间达38个月,目前仍在随访中。有文献报道,病理Hans分型GCB来源患者预后较non-GCB来源患者预后好,5年OS约为90%[18]。本组中GCB患者6例,2例因疾病进展死亡;non-GCB患者9例,1例因疾病进展死亡,1例因感染死亡,两组患者的预后差异无统计学意义,可能与本组病例数较少,随访时间较短有关。
综上所述,PB-DLBCL是一种少见且预后较好的恶性肿瘤,临床表现以局部疼痛为主,病变多为累及颌骨、长骨和扁骨的溶骨性病变,对于影像学怀疑的骨肿瘤性病变患者,应早期行病理学检查,诊断PB-DLBCL时需将病理形态学改变、免疫组化及分子检测相结合以提高诊断准确性,早期明确诊断并进行及时规范化治疗可有效提高患者的生存率。
猜你喜欢
传染病信息(2022年3期)2022-07-15
中国典型病例大全(2022年12期)2022-05-13
新医学(2022年4期)2022-04-23
临床与实验病理学杂志(2021年7期)2021-12-03
保健医苑(2021年7期)2021-08-13
现代临床医学(2021年4期)2021-07-31
抗癌(2021年2期)2021-07-09
医学食疗与健康(2021年27期)2021-05-13
饮食与健康·下旬刊(2017年4期)2017-05-26
中国民族民间医药·下半月(2016年8期)2016-10-24
