
乙炔三聚环化制苯反应机理的理论研究
2022-01-17 08:21余盛萍郭晓蕊石佳玉
西南民族大学学报(自然科学版) 2021年2期
余盛萍,郭晓蕊,石佳玉
(西南民族大学化学与环境学院 国家民委化学基础重点实验室, 四川 成都 610041)
乙炔和苯都是化工生产中重要有机原料,在各化工领域中具有重要作用.Berthelot等人[1]在1866年首次发现可以通过乙炔三聚环化生成苯.随后发现在催化C2H2三聚环化反应中过渡金属Ni, Co, Pd, Cr, Rh, Fe, Zr, Nb, Ir和 Ta等形成的过渡金属复合物、金属负载催化剂和金属氧化物催化剂[2-6]等都能表现出很好的催化性能,研究表明其中Pd单原子催化剂对炔烃加氢反应具有很高的活性和热稳定性.此外,MgO晶体结构相对于分子晶体具有较高的硬度和熔沸点,在一般条件下性质较为稳定,可以作为良好的催化剂载体.Rösch等人[7]报道用质量分离软着陆技术将Pdn团簇(1n30)负载在MgO(100)面上可以在低温(300 K)的条件下催化乙炔三聚制苯.Yang等人[8]采用密度泛函方法研究了FeO催化乙炔三聚环化气相反应机理.本文Pd单原子分别负载到2×2×2和3×3×3的MgO晶体载体上得到两种单原子催化剂PdMg4O4和PdMg14O13,对比研究这两种单原子催化剂对乙炔催化三聚成环反应中各驻点(包括反应物、中间体、过渡态和产物)的影响,以揭示其反应机理.
1 计算方法
密度泛函理论应用于Pd单原子催化的理论研究[9-11]表明在结构优化和计算光电子能谱等方面皆可获得较好的结果,采用B3LYP[12-13]方法和赝势基组Lanl2dz[14-15]能得到更好的结果.在B3LYP/Lanl2dz水平下优化,得到了稳定的PdMg4O4和PdMg14O13两种载体结构.本文首先在B3LYP/Lanl2dz水平下分别优化这两种单原子催化剂催化下乙炔三聚环化成苯反应中单重态势能面上的各驻点的结构, 包括反应物、过渡态、中间体和产物,并在相同水平下计算了驻点的频率,采用內禀坐标( IRC)方法验证所有过渡态.在此基础上进行了自然键轨道(NBO)[16]分析,计算各物种的相对能量以及原子的自然电荷,全部计算由Gaussian 16程序包[17]完成.
2 结果与讨论
2.1 单原子催化剂PdMg4O4催化乙炔三聚环化制苯
乙炔三聚环化制苯反应的吉布斯自由能变化值为-531.2 kJ/mol,与前人计算吉布斯自由能变化值为-533.9 kJ/mol[18]相当吻合.因此,该反应在热力学上是能进行的.
图1和图2分别绘出了单原子催化剂PdMg4O4催化乙炔三聚环化成苯过程中各驻点的稳定结构和反应能级示意图.首先,PdMg4O4催化剂吸附第一个乙炔分子结合形成复合物IM1(PdMg4O4C2H2).第二步,IM1与第二个乙炔分子吸附生成复合物IM2 (C2H2PdMg4O4C2H2).第三步,IM2经过五元环过渡态TS1,环化过程中生成新的C―C键,发生氧化加成生成含Pd的五元环复合物IM3(cyc-PdMg4O4C4H4).第四步,IM3吸附第三个乙炔分子,生成分子复合物IM4(C2H2PdMg4O4C4H4).第五步,IM4经过过渡态TS2,环化生成新的C―C键,同时Pd―C键断裂,使碳环闭合,生成中间体IM5(PdMg4O4C6H6).最后,IM5释放出苯分子.

图1 PdMg4O4催化的各驻点的稳定结构(键长单位为Å)Fig. 1 The stable structure of each stagnation point with PdMg4O4 (The bond length is in Å)

图2 PdMg4O4催化乙炔三聚环化成苯的反应能级示意图(能量△E单位为KJ/mol)Fig. 2 Potential energy profile of cyclization of acetylene to benzene catalyzed by PdMg4O4 (△E is in KJ/mol)
如图1所示, 中间体IM1中的Pd―O键长为2.083 Å, 较单个PdMg4O4的Pd―O键长2.042 Å有所增长, 这表明C2H2与PdMg4O4形成中间体IM1后Pd―O键得到活化.在IM2→TS1→IM3过程中,部分Pd―C键由最初的2.048 Å增长至2.138 Å,到最终的2.887 Å,而部分C―C键由最初的3.104 Å,逐渐减短至2.134 Å,到最终的1.452 Å;IM4→TS2→IM5过程中,部分Pd―C键由最初的2.029 Å增长至2.054 Å,到最终的3.608 Å,而部分C―C键由最初的3.066 Å,逐渐减短至2.717 Å,到最终的1.434 Å,这些键长的变化说明催化过程中Pd―C键逐渐断裂、C―C键逐渐形成.
如图2所示,乙炔三聚环化制苯反应放热-632.0 kJ/mol,TS1和TS2的能垒分别为105.4 KJ/mol和196.9 KJ/mol.能量最高点为反应物PdMg4O4+3C2H2的总能量,其后整体的势能曲线呈下降趋势,能量最低点为中间体IM5,处于-730.5 kJ/mol的深势阱中.其中,TS2是关键过渡态,其能垒最高,是整个反应的速率控制步骤.
2.2 单原子催化剂PdMg14O13催化乙炔三聚环化制苯
图3和图4分别绘出了PdMg14O13催化乙炔三聚环化生成苯过程中各驻点的稳定结构和反应能级示意图.其反应过程类似于PdMg4O4催化.中间体IM6中的Pd―O键键长比IM1中的短,这表明C2H2与PdMg14O13形成中间体 IM6中Pd―O键更容易被活化.同样,Pd―C键和C―C键的变化说明催化过程中Pd―C键逐渐断裂和C―C键逐渐形成.
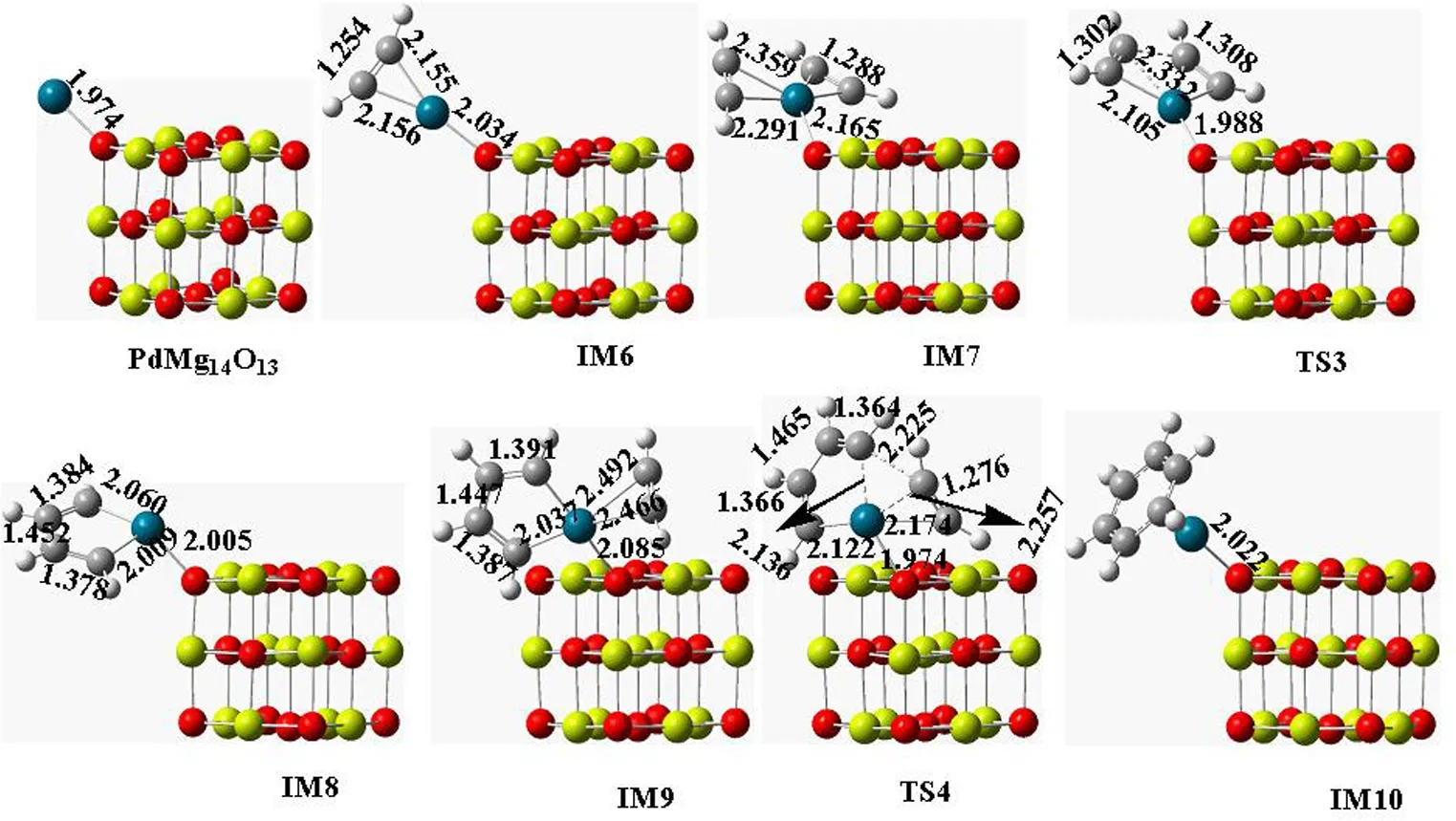
图3 PdMg14O13催化的各驻点的稳定结构(键长单位为Å)
通过对比图2和图4的反应能级示意图,催化剂不同,其反应发出的热相同,TS3和TS4的能垒分别为100.5 KJ/mol和106.0 KJ/mol,分别比TS1和TS2的能垒小.PdMg14O13单原子催化剂催化乙炔三聚成环时决速步骤的反应能垒较PdMg4O4单原子催化剂的对应的过渡态能垒低,这表明选取3×3×3模型PdMg14O13作为单原子催化剂具有更好的催化效果,反应更容易进行.类似于PdMg4O4催化,其中能量最高点也为反应物的能量总和,整体的能量数值呈下降趋势,能量最低点为中间体IM10(PdMg14O13C6H6),处于-2069.9 KJ/mol的深势阱中.该反应的速控步骤是过渡态TS4,为关键过渡态,是反应的决速步骤.
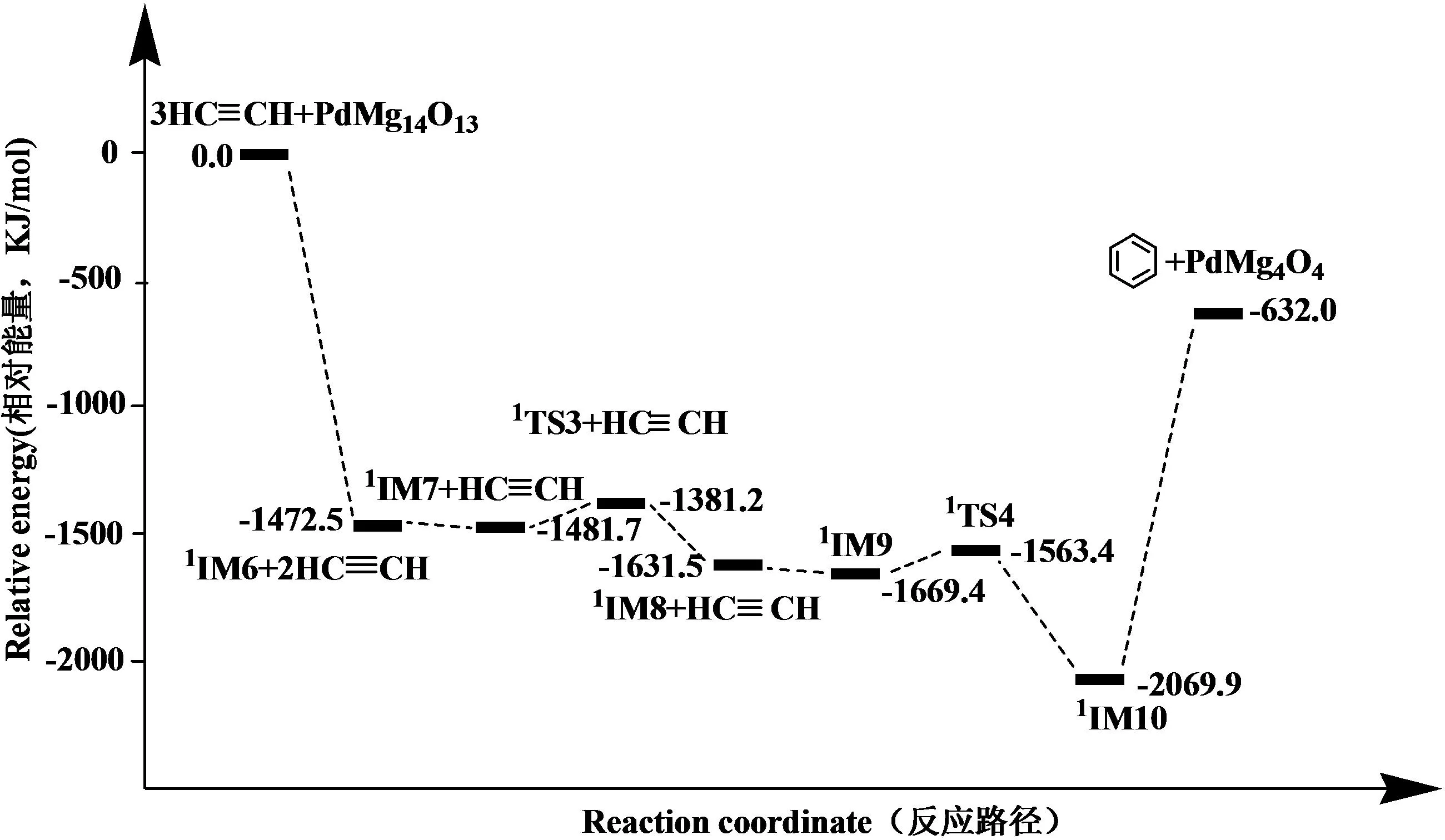
图4 PdMg14O13催化乙炔三聚环化成苯的反应能级示意图(能量△E的单位为KJ/mol)Fig. 4 Potential energy profile of cyclization of acetylene to benzene catalyzed by PdMg14O13 (△E is in KJ/mol)
2.3 电荷分析
表1列出了各驻点中PdMg4O4、PdMg14O13和CnHn各部分的自然电荷.可以发现非常重要的电子转移规律,如表1所示,CnHn的净电荷为负值,而催化剂PdMg4O4的净电荷为正值,电子从金属负载催化剂PdMg4O4流向CnHn,即金属负载催化剂是电子供体,提供电子给CnHn.当n从2变化到4最后增加到6时,CnHn的自然电荷总体上呈增多的趋势,这是由于乙炔分子受到Pd原子的金属化,发生亲核加成,导致吸电子作用增强,因此转移的电子数增多.同理,当催化剂为PdMg14O13时,n从2到6时,CnHn的自然电荷总体上也呈增多的趋势.
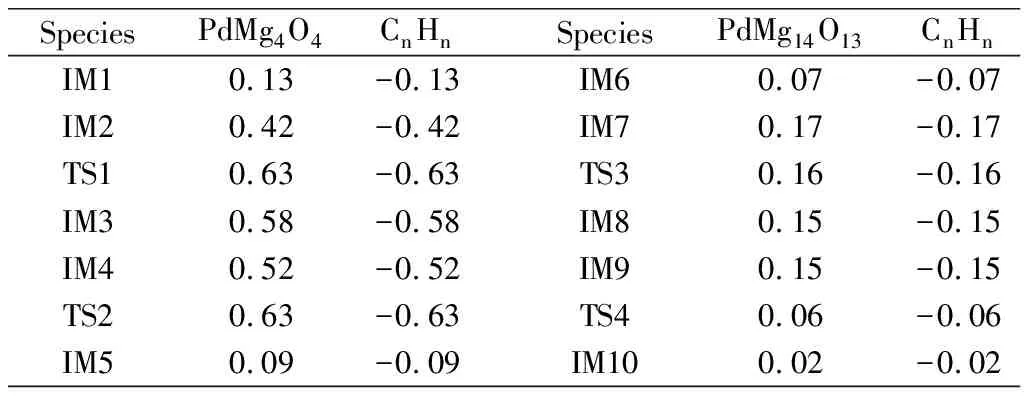
表1 在B3LYP/Lanl2dz水平下反应物种中各部分原子的自然电荷Table 1 Natural charge of some part atoms of species at B3LYP / Lanl2dz level
3 结论
在B3LYP/Lanl2dz水平下,我们对比研究了单原子催化剂PdMg4O4和PdMg14O13催化乙炔三聚制苯的反应机理,研究表明:第一,当金属负载催化剂催化C2H2三聚环化的反应时吉布斯自由能为-531.2 KJ/mol,这在热力学上是有利的.第二,CnHn的净电荷为负值,而Pd单原子催化剂的净电荷为正值,说明电子从Pd单原子催化剂流向CnHn,即Pd单原子催化剂是电子供体,CnHn是电子受体.第三,Pd/MgO作为乙炔催化反应的催化剂时,具有良好的活性及热稳定性,PdMg14O13催化剂模型催化乙炔三聚成环时决速步骤TS4的反应能垒较PdMg4O4催化剂模型的决速步骤TS2的能垒低,这表明选取3×3×3模型PdMg14O13作为催化剂具有更好的催化效果.
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
商品与质量(2021年31期)2021-11-23
疯狂英语·新阅版(2019年6期)2019-09-10
科普创作(2018年1期)2018-11-30
山东工业技术(2018年9期)2018-05-26
中学生数理化·高二版(2017年3期)2017-07-07
党的生活·党员电教与远程教育(2016年3期)2016-02-26
湖北教育·综合资讯(2016年1期)2016-02-19
源流(2015年8期)2015-09-16
股市动态分析(2015年12期)2015-09-10
