
抗高血压药物维拉帕米的合成
2022-01-12 12:51:08李旻玥宋荣聪
合成化学 2021年12期
李旻玥, 宋荣聪, 任 杰, 胡 昆
(常州大学 药学院,江苏 常州 213164)
高血压(Hypertension,HTN)是世界范围内最普遍的慢性病,其特征为全身动脉血压升高,可伴有心、脑、肾等器官造成损伤的临床综合征,也是心血管疾病[1](CVD)的关键危险因素之一。高血压发病的主要因素或假说有以下几种:年龄[2]、遗传[3]、环境因素[4]、交感神经系统学说[5]、血管反应性学说[6]、肾微血管疾病假说[7]、尿酸因子学说[8]等。
维拉帕米(Verapamil)是由德国Knoll制药公司原研,1963年以商品名Isoptin[9]在德国上市。维拉帕米可以抗心律失常和抗心绞痛,广泛用于原发性高血压[10]的治疗。与高血压的其他药物相比,维拉帕米具有显著优点[11-12]:(1)降压作用强,且能持久有效地控制血压水平[13];(2)副作用小,安全方便[14];(3)显著较少脑卒中事件。因此维拉帕米成为治疗高血压最有价值的药物。
本文对国内外维拉帕米合成方法做了总结,主要有以下3种:(1)BASH公司[15]报道了一条维拉帕米的合成路线,该路线以愈创木酚为起始原料,经O-甲基化、氯甲基化、氰化、C-烷基化得到关键中间体α-异丙基-3,4-二甲氧基苯乙腈,再与N-(γ-氯丙基)-3,4-二甲氧基苯乙胺直接缩合得到去甲基化维拉帕米,最后进行N-甲基化反应得到目标产物维拉帕米,总收率为11.3%。该路线的起始原料较为便宜,但在3,4-二甲氧基苯乙腈的合成过程中需用到剧毒试剂氰化钠。此外,在合成中间体3,4-二甲氧基苯乙胺时,需用到高压条件,对设备要求较高。(2)德国Knoll公司[16]提出了一条同样是以愈创木酚为起始原料的合成路线。该路线在合成α-异丙基-3,4-二甲氧基苯乙腈时与直接合成法类似,都是经过O-甲基化、氯甲基化、氰化、C-烷基化四步得到,再经过C-烷基化、脱保护得到关键中间体α-(γ-丙基醛)-α-异丙基-3,4-二甲氧基苯乙腈,最后与3,4-二甲氧基苯乙胺经醛胺缩合、N-甲基化、成盐反应得到终产物盐酸维拉帕米,总收率为11.6%。此路线在合成3,4-二甲氧基苯乙腈时,同样不可避免地用到了剧毒试剂氰化钠。此外,在两次C-烷基化的过程中需用到大量的NaNH2使得后处理繁琐,且对环境和安全都存在着隐患,而且3-氯丙醛二乙缩醛生产繁琐,难以实现大规模化。(3)Lothar等[17]提出了一条以α-异丙基-3,4-二甲氧基氯化苄为起始原料的合成路线。该路线经氰化、Michael加成、腈胺缩合得到终产品维拉帕米,总收率为42.8%。该路线起始原料较为昂贵且不易得到,在氰化过程中还是要用到剧毒试剂氰化钠,此外合成终产品时需用到高压条件,对设备要求较高,增加了生产成本。
本文设计了一条新的维拉帕米的合成路线。以3,4-二甲氧基苯乙酸(8)为原料,经过酰化反应生成3,4-二甲氧基苯乙酰胺(7);7经脱水反应得到3,4-二甲氧基苯乙腈(6);6与2-溴丙烷经烷基化反应生成中间体α-异丙基-3,4-二甲氧基苯乙腈(5);再以3,4-二甲氧基苯乙酸(8)为原料,经酰化反应生成N-甲基-3,4-二甲氧基苯乙酰胺(4);4经还原反应生成N-甲基-3,4-二甲氧基苯乙胺(3);3与1-溴-3氯丙烷经烷基化反应生成中间体N-(γ-氯丙基)-N-甲基-3,4-二甲氧基苯乙胺(2),最后中间体2和α-异丙基-3,4-二甲氧基苯乙腈(5)经缩合反应生成最终目标产物维拉帕米(1, Scheme 1),总收率为38.6%,其结构经1H NMR和13C NMR确证。该合成路线主要有以下创新点:(1)起始原料及使用的试剂廉价较为易得;(2)操作简便,反应条件温和,避免使用剧毒试剂,且后处理简便,不用柱层析;(3)各步反应收率较高,重复性好。

Scheme 1
1 实验部分
1.1 仪器与试剂
Bruker Avance III 400/500 MHz型核磁共振仪(TMS为内标,DMSO或CDCl3为溶剂)。
所用试剂均为分析纯。
1.2 合成
(1)7的合成
向250 mL圆底烧瓶中加入3,4-二甲氧基苯乙酸(8)10.00 g(50.97 mmol),然后加入50 mL氯化亚砜将其溶解,回流反应1 h。减压蒸干氯化亚砜得黄色油状液体,然后用乙腈20 mL将其溶解,逐滴加入50 mL冰浴下的氨水中,30 min后反应结束。减压蒸干乙腈,有白色固体析出,抽滤,滤饼用水(3×50 mL)洗涤,真空干燥得到白色固体79.29 g,收率93.4%;1H NMR(400 MHz, CDCl3)δ: 6.98~6.69(m, 3H), 6.04(s, 1H), 5.53(s, 1H), 3.89(s, 3H), 3.87(s, 3H), 3.52(s, 2H);13C NMR(100 MHz, CDCl3)δ: 174.16, 149.25, 148.35, 127.33, 121.57, 112.35, 111.50, 55.94, 55.91, 42.94。
(2)6的合成
向250 mL圆底烧瓶中加入79.00 g(46.11 mmol),用甲苯50 mL将其溶解,然后加入五氧化二磷13.11 g(92.22 mmol),回流反应1 h。加入水50 mL,用乙酸乙酯萃取(50 mL×3),合并有机相,饱和食盐水(3×150 mL)洗涤滤液,收集有机相,无水硫酸镁干燥,抽滤,减压蒸干溶剂得黄色油状物67.29 g,收率89.2%;1H NMR(400 MHz, CDCl3)δ: 7.15~6.57(m, 3H), 3.91(s, 3H), 3.89(s, 3H), 3.71(s, 2H);13C NMR(100 MHz, CDCl3)δ: 149.39, 148.80, 122.14, 120.24, 118.19, 111.45, 110.92, 55.99, 55.98, 23.24。
(3)5的合成
将67.00 g(39.50 mmol)加入到100 mL圆底烧瓶中,用DMSO 5 mL溶解,然后依次加入TEBA 0.52 g(1.98 mmol), 50%氢氧化钠溶液20 mL, 2-溴丙烷7.33 g(59.25 mmol)50 ℃反应2 h。加入水50 mL,用乙酸乙酯萃取(3×50 mL),合并有机相,用饱和食盐水(3×150 mL)洗涤,无水硫酸镁干燥,抽滤,滤饼减压蒸干溶剂,残余物用50%乙醇水溶液重结晶得白色固体57.64 g,收率88.4%;1H NMR(400 MHz, CDCl3)δ: 7.06~6.60(m, 3H), 3.92(s, 3H), 3.90(s, 3H), 3.61(d,J=6.5 Hz, 1H), 2.11(dt,J=13.3, 6.7 Hz, 1H), 1.06(d,J=6.7 Hz, 6H);13C NMR(100 MHz, CDCl3)δ: 149.13, 148.71, 127.36, 120.26, 120.10, 111.16, 110.75, 56.00, 55.96, 44.77, 33.87, 20.78, 18.97。
(4)4的合成
将3,4-二甲氧基苯乙酸(8)10.00 g(50.97 mmol)置于250 mL圆底烧瓶中,加入甲醇50 mL溶解,然后加入浓H2SO4(1 mL)回流反应4 h。减压蒸干甲醇得黄色油状物,后用甲胺的醇溶液(50 mL)将其溶解,室温反应5 h。减压蒸干溶剂,加入饱和NaHCO3水溶液(50 mL),用乙酸乙酯萃取(3×50 mL),合并有机相,用饱和食盐水(3×150 mL)洗涤,无水硫酸镁干燥,抽滤,滤饼减压蒸干溶剂得黄色固体49.52 g,收率89.3%;1H NMR(400 MHz, CDCl3)δ: 6.99~6.64(m, 3H), 3.84(s, 3H), 3.82(s, 3H), 3.46(s, 2H), 2.72(d,J=4.9 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 172.00, 149.18, 148.23, 127.42, 121.61, 112.49, 111.48, 55.90, 55.86, 43.19, 26.45。
(5)3的合成
将NaBH46.56 g(172.06 mmol)置于250 mL圆底烧瓶中,加入干燥四氢呋喃50 mL溶解,冰浴下缓慢滴加BF3·Et2O 24.42 g(172.06 mmol)搅拌30 min,然后加入69.00 g(43.01 mmol)的干燥四氢呋喃溶液30 mL,室温反应6 h。加入适量甲醇淬灭反应,减压旋干反应液,加入2 M盐酸调pH至2~3,用乙酸乙酯(3×30 mL)萃取,合并水相,加入20%NaOH溶液调pH至12~13,用乙酸乙酯(3×50 mL)萃取,合并有机相,用饱和食盐水(150 mL×3)洗涤,无水硫酸镁干燥,抽滤,滤液减压蒸干溶剂得黄色油状液体 7.53 g,收率89.5%;1H NMR(400 MHz, CDCl3)δ: 6.77(dd,J=23.2 Hz, 7.8 Hz, 3H), 3.87(s, 3H), 3.85(s, 3H), 2.91~2.66(m, 4H), 2.43(s, 3H), 2.02(s, 1H);13C NMR(100 MHz, CDCl3)δ: 148.88, 147.40, 132.49, 120.56, 111.92, 111.27, 55.91, 55.82, 53.23, 36.27, 35.68。
(6)2的合成
将36.00 g(30.73 mmol)置于250 mL圆底烧瓶中,用DMF 30 mL溶解,然后依次加入碳酸钠13.0 g(122.91 mmol)、 1-溴-3-氯丙烷7.32 g(46.10 mmol),室温反应8 h。加入水30 mL,加入2 M盐酸调pH至2~3,用乙酸乙酯(3×50 mL)萃取,合并水相,加入20% NaOH溶液调pH至12~13,用乙酸乙酯(3×50 mL)萃取,合并有机相,用饱和食盐水(3×150 mL)洗涤,无水硫酸镁干燥,抽滤,滤液减压蒸干溶剂得黄色油状液体26.91 g,收率82.5%;1H NMR(400 MHz, CDCl3)δ: 7.00~6.60(m, 3H), 3.89(s, 3H), 3.87(s, 3H), 3.59(t,J=6.5 Hz, 2H), 2.80~2.69(m, 2H), 2.67-2.52(m, 4H), 2.31(s, 3H), 1.95(p,J=6.7 Hz, 2H);13C NMR(100 MHz, CDCl3)δ: 148.79, 147.29, 133.06, 120.50, 111.99, 111.21, 59.83, 55.93, 55.85, 54.37, 43.26, 42.27, 33.49, 30.36。
(7)1的合成
将55.00 g(22.80 mmol)置于100 mL圆底烧瓶中,用20 mL甲苯溶解,然后依次加入TEBA 0.32 g(1.14 mmol)、 KOH粉末10.20 g(182.40 mmol),最后在45 min内滴加25.63 g(20.52 mmol)的甲苯溶液10 mL,氮气保护下90 ℃反应4 h。加入水30 mL,加入2 M盐酸调pH至2~3,用乙酸乙酯萃取(3×50 mL),合并水相,加入20% NaOH溶液调pH至12~13,用乙酸乙酯(3×50 mL)萃取,合并有机相,用饱和食盐水(3×150 mL)洗涤,无水硫酸镁干燥,抽滤,滤液减压蒸干溶剂得黄色油状液体15.40 g,收率52.4%;1H NMR(500 MHz, CDCl3)δ: 7.01~6.62(m, 6H), 3.90(s, 3H), 3.89(s, 3H), 3.88(s, 3H), 3.87(s, 3H), 2.75~2.61(m, 2H), 2.51(t,J=7.5 Hz, 2H), 2.37(ddq,J=18.2 Hz, 12.3 Hz, 6.5 Hz, 5.9 Hz, 2H), 2.20(s, 3H), 2.16 -2.04(m, 2H), 1.84(td,J=13.5 Hz, 13.0 Hz, 4.4 Hz, 2H), 1.57(tt,J=12.0 Hz, 6.0 Hz, 1H), 1.20(d,J=6.7 Hz, 3H), 0.80(d,J=6.7 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 148.96, 148.78, 148.22, 147.26, 133.05, 130.69, 120.49, 118.66, 111.97, 111.17, 111.04, 109.51, 59.54, 56.96, 55.98, 55.92, 55.89, 55.84, 53.39, 42.02, 37.95, 35.64, 33.27, 23.43, 19.00, 18.63。
2 结果与讨论
2.1 6的反应条件优化
该反应为酰胺脱水成氰基的反应,相关文献[18-19]发现,不同的脱水试剂和反应溶剂对实验的收率有一定的影响。为了能找到合适的反应条件,考察了不同脱水试剂和反应溶剂对反应收率的影响。
(1) 脱水剂
以化合物75.1 mmol为原料,分析了不同脱水试剂对6收率的影响,所有收率均为3次平行实验的平均值,结果见表1。由表1可知,当选择五氧化二磷为脱水试剂时,产物6的收率最高(89.2%)。
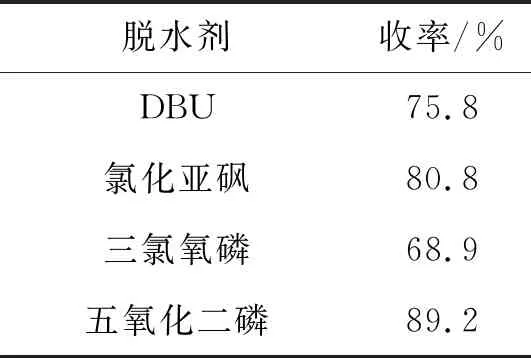
表1 不同脱水试剂下6的收率
(2) 反应溶剂
以化合物75.1 mmol为原料,2.0 eq.的五氧化二磷为脱水试剂,反应溶剂的用量为15 mL,实验不同反应溶剂对产物6收率的影响,所有收率均为3次平行实验的平均值,实验结果可见表2。由表2可知,当使用甲苯为溶剂时,产物6的收率最高。

表2 不同溶剂下6的收率
2.2 合成5的反应条件优化
该反应为C-烷基化反应,主要指有机化合物中碳原子上引入烷基的反应,常用的烷基化试剂有卤代烃、醇、烯烃等。影响该反应收率的主要因素有烷基化试剂、有机物自身结构、催化剂、反应溶剂、反应时间等。主要考察不同催化剂和反应时间对反应收率的影响。
(1) 催化剂
以化合物65.6 mmol为原料,1.5 eq. 2-溴丙烷为烃化剂,氢氧化钠水溶液(1/1)为碱,二甲亚砜3 mL为溶剂,反应温度为50 ℃,反应时间为2 h,实验了催化剂对5收率的影响,所有收率均为3次平行实验的平均值,实验结果见表3。由表3可知,TEBA为催化剂时,5的收率最高。

表3 不同催化剂下5的收率
(2) 反应时间
以化合物65.6 mmol为原料,1.5 eq. 2-溴丙烷为烃化剂,0.05 eq. TEBA为催化剂,氢氧化钠溶液(1/1)为碱,氯化亚砜3 mL为溶剂,反应温度为50 ℃,实验不同反应时间对反应收率的影响,所有收率均为三次平行实验的平均值,实验结果可见表4。由表4可知,当反应时间为2 h时,收率趋于最大值,当再延长反应时间后,收率承下降趋势,分析原因可能是产物5自己水解导致,因此该反应的最优时间为2 h。

表4 不同反应时间下5的收率
2.3 合成3的反应条件优化
该反应为酰胺的还原反应,在一些条件下,常伴有碳—氮键的断裂生成醛,故还原剂和原料配比对收率起到关键的作用,为了寻找最佳的反应收率,实验了不同还原剂和原料配比[r=n(硼氢化钠-三氟化硼乙醚/n(4)]对反应收率的影响。
(1) 还原剂[20-21]
以化合物44.8 mmol为原料,考察不同还原剂对3收率的影响,所有收率均为三次平行实验的平均值,实验结果见表5。从表5可以看出,当选择硼氢化钠/三氟化硼乙醚为还原剂时,反应的效果最好,因此该反应最好的还原剂为硼氢化钠/三氟化硼乙醚体系。

表5 不同还原剂下3的收率
(2) 原料配比
以化合物44.8 mmol为原料,硼氢化钠/三氟化硼乙醚为还原剂,实验了不同原料配比对3反应收率的影响,所有收率均为三次平行实验的平均值,实验结果见表6。从表6可以看出,当原料配比为4时,产品的收率趋于稳定,再增加原料配比对收率影响不大,故选择原料配比4为最佳的反应条件。
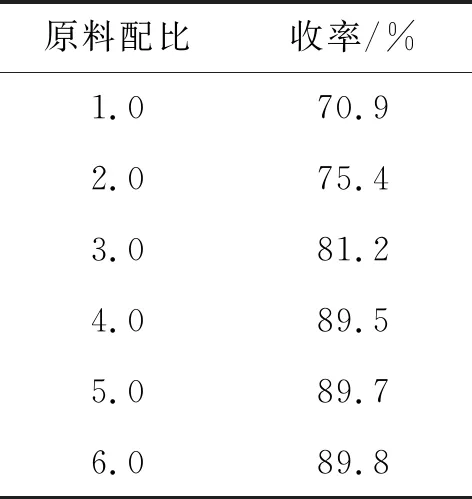
表6 不同原料配比对3收率的影响
以3,4-二甲氧基苯乙酸为原料制得关键中间体α-异丙基-3,4-二甲氧基苯乙腈(5);再以3,4-二甲氧基苯乙酸为原料,经酰化、还原、N-烷烃化反应得到N-(γ-氯丙基)-N-甲基-3,4-二甲氧基苯乙胺(2);2与5经过缩合反应合成了最终产物维拉帕米,并对部分中间体的合成条件进行了优化,总收率为25.4%。与已有合成路线相比,该合成路线起始原料相对廉价易得、操作简便、反应条件温和、后处理简单,各步反应收率较高,经多次平行反应证明重复性好,具有较好的应用前景。
猜你喜欢
江南诗(2023年6期)2023-12-08 05:17:24
金沙江文艺(2022年1期)2022-02-04 10:15:16
化学教与学(2021年12期)2021-02-18 01:16:58
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24 21:02:17
南方周末(2014-09-25)2014-09-25 01:43:01
中国药业(2014年21期)2014-05-26 08:56:28
中国药业(2014年19期)2014-05-17 03:11:57
计算物理(2014年1期)2014-03-11 17:00:14
中外医疗(2013年16期)2013-02-01 21:49:07
中国合理用药探索(2011年3期)2011-03-20 16:30:16
