
超支化极性官能团化烯烃齐聚物的合成及表征
2022-01-12 12:51严正鹏李帅康代胜瑜
合成化学 2021年12期
严正鹏, 李帅康, 代胜瑜
(安徽大学 物质科学与信息技术研究院,安徽 合肥 230601)
高分子量聚烯烃如高密度聚乙烯和线性低密度聚乙烯作为热塑性材料广泛应用于我们日常生产和生活中[1-2],有关其合成的研究已引起学术界和工业界的高度重视,并取得了重大进展[3-4]。与此不同的是,低分子量齐聚烯烃的研究则相对较少。相比高分子量聚烯烃,低分子量齐聚烯烃表现出的特有性质使其在特殊场合具有重要应用价值,比如作为润滑油和表面活性剂[5-6]。因此,发展高效合成低分子量烯烃齐聚物的方法具有重要意义。
目前工业上主要采用特殊结构茂金属催化剂或中性镍催化剂来制备乙烯齐聚物,产物主要为线性齐聚物,若要制备支化甚至超支化的烯烃齐聚物往往需要以价格昂贵的α-烯烃为原料。近些年,后过渡链行走金属催化剂的发展为制备这类支化和超支化乙烯齐聚物提供了新的思路和方法,这些催化剂能够以乙烯为单一原料制备支化甚至超支化的烯烃齐聚物[7-9]。其中“链行走”阳离子α-二亚胺钯催化体系因其可能实现支化极性官能团化乙烯齐聚物的有效合成而受到广泛关注[10-22]。一般而言,邻位芳基大位阻取代基有利于延缓聚合过程中的链转移和保护催化活性中心,从而提高所得聚乙烯分子量和聚合活性[23-24]。因此,具有邻位芳基大位阻取代的二亚胺钯催化剂能够催化制备出高分子量高支化度的聚乙烯和极性官能团化聚乙烯。这类催化剂具有优异的链行走和催化乙烯与极性单体共聚的能力。在此基础上为进一步获得低分子量的超支化乙烯齐聚物,科研工作者设计了小位阻轴向取代或骨架衍生的二亚胺钯催化剂。叶等人发现无位阻乙二醛骨架的二亚胺钯催化剂可催化乙烯聚合制备高支化的低分子量聚乙烯(Chart 1A)[25]。但因缺少大位阻骨架的保护,这类催化剂很容易失活,导致该体系稳定性和活性较低。此外,Milani 等人开发了轴向小位阻不对称苊醌骨架的二亚胺体系,该体系能催化乙烯与丙烯酸甲酯(MA)共聚制备高极性单体插入比的高支化极性官能团化乙烯齐聚物(Chart 1B)[26]。然而同样存在的问题是,没有大位阻轴向取代基的保护,这类体系的稳定性和聚合活性也较低。
为解决上述问题,课题组曾开发了一类单边大位阻取代的不对二亚胺钯催化体系,该单边大位阻取代基对维持催化剂稳定性和聚合活性具有重要作用。利用该钯催化体系可催化乙烯与极性单体齐聚制备超支化的极性官能团化的乙烯齐聚物(Chart 1C)[27]。然而,该体系共聚极性单体能力较差,所得共聚物插入比较低。因此,进一步开发新型的催化体系,以解决支化极性官能团化乙烯齐聚物制备过程中稳定性差和插入率低的问题,显得十分重要。近期高海洋课题组和我们在经典吡啶亚胺体系基础上(Chart 1D)[28]利用大位阻二芳基甲基苯胺合成一类新型吡啶亚胺钯催化剂(Chart 1E-F),该体系在催化过程中可保持高活性和高稳定性,催化乙烯齐聚制备超支化乙烯齐聚物[29-31]。此外,这类催化剂能够催化乙烯与极性单体共聚,得到高插入比的极性官能团化乙烯齐聚物[32]。在此工作基础上,本文设计合成了了一类刚柔并济的吡啶亚胺钯催化剂,并将其应用于乙烯或丙烯齐聚以及乙烯或丙烯与MA共齐聚中(Scheme 1)。希望利用该体系制备超支化乙烯或丙烯齐聚物和具有高插入比的极性官能团化超支化乙烯或丙烯齐聚物。

Chart 1

Chart 2
1 实验部分
1.1 仪器与试剂
JNM-ECZ600R/Z400R型核磁共振仪(CDCl3为溶剂);ESI/APCI型质谱仪;Elementar型元素分析仪。
所用试剂均为分析纯。
1.2 吡啶亚胺配体及其钯配合物合成
(1) 大位阻芳胺的合成(Scheme 2)
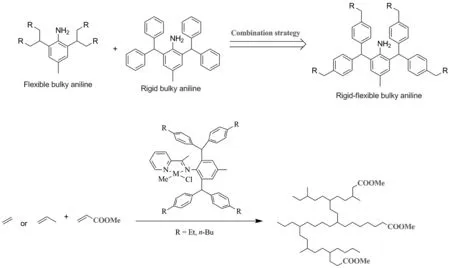
Scheme 1
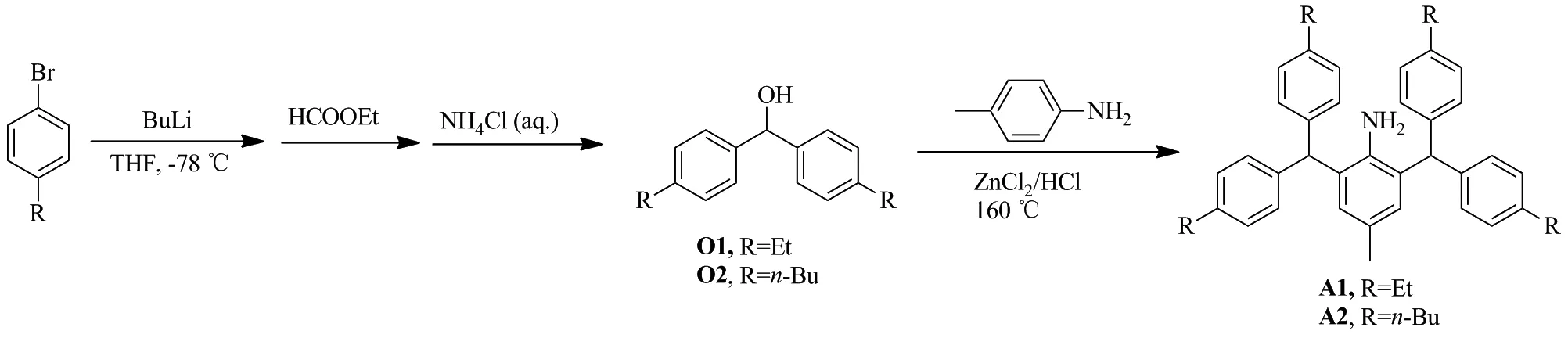
Scheme 2
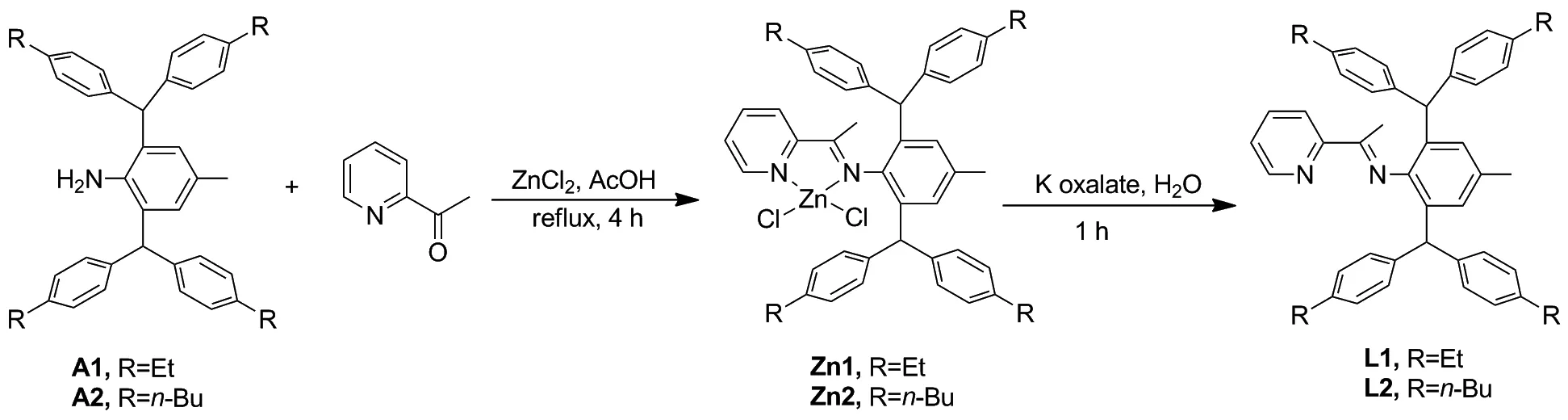
Scheme 3
将芳基溴80 mmol(2.0 eq.)在-78 ℃条件下溶于300 mL的四氢呋喃中,然后将33 mL丁基锂(2.5 mol/L)缓慢注射到上述混合物中。反应30 min后,将甲酸乙酯40 mmol加入上述反应物,反应物接着在-78 ℃条件下反应0.5 h。之后将反应体系移到室温进行反应5 h。反应结束时加入10 mL氯化铵水溶液进行淬灭,旋蒸除去大部分有机溶剂,加入150 mL水,用3×100 mL二氯甲烷溶剂萃取。收集有机相溶剂,用无水硫酸镁干燥。减压蒸发溶剂,产物通过柱色谱方式分离。
O1: 6.5 g,收率68%;1H NMR(600 MHz, CDCl3)δ: 7.30(d,J=8.1 Hz, 4H, Ar-H), 7.18(d,J=8.0 Hz, 4H, Ar-H), 5.79(s, 1H, CHAr2), 2.65(q,J=7.6 Hz, 4H, CH2CH3), 2.29(s,br, 1H, OH), 1.24(t,J=7.6 Hz, 6H,CH2CH3);13C NMR(151 MHz, CDCl3)δ: 143.59, 141.47, 128.04, 126.63, 76.09(CHAr2), 28.64(CH2CH3), 15.67(CH2CH3)。
O2: 8.6 g, 收率43%;1H NMR(600 MHz, CDCl3)δ: 7.28(d,J=8.0 Hz, 4H, Ar-H), 7.15(d,J=8.0 Hz, 4H, Ar-H), 5.78(s, 1H, CHAr2), 2.65~2.57(m, 4H, CH2CH2CH2CH3), 2.27(s,br, 1H, OH), 1.65~1.55(m, 4H, CH2CH2CH2CH3), 1.42~1.32(m,J=7.9 Hz, 4H,CH2CH2CH2CH3), 0.94(t,J=7.3 Hz, 6H, CH2CH2CH2CH3);13C NMR(151 MHz, CDCl3)δ: 142.25, 141.40, 128.58, 126.56, 76.11(CHAr2), 35.42(CH2CH2CH2CH3), 33.72(CH2CH2CH2CH3), 22.48(CH2CH2CH2CH3), 14.05(CH2CH2CH2CH3)。
将二芳基甲醇20.0 mmol(2.0 eq.)和对甲基苯胺1.07 g(10 mmol, 1.0 eq.)的混合物加热至120 ℃。此时缓慢加入无水氯化锌0.68 g(5 mmol, 0.5 eq.)和浓盐酸0.6 mL(37% in H2O, 1.0 eq.)的混合液,滴加完毕后将反应温度升高至160 ℃。反应30 min后,将反应体系冷却至室温,加入200 mL二氯甲烷溶解。混合物用水(3×100 mL)洗涤,无水硫酸镁干燥后,溶液浓缩至20 mL。加入200 mL乙醇沉淀出大量固体,过滤,滤饼用乙醇(3×20 mL)洗涤。
A1: 4.58 g,收率83%;1H NMR(600 MHz, CDCl3)δ: 7.12(d,J=8.0 Hz, 8H, Ar-H), 7.03(d,J=8.1 Hz, 8H, Ar-H), 6.43(s, 2H, Ar-H), 5.41(s, 2H, CHAr2), 3.32(s,br, 2H, NH2), 2.65(q,J=7.6 Hz, 8H, CH2CH3), 2.06(s, 3H, CH3), 1.25(t,J=7.6 Hz, 12H, CH2CH3);13C NMR(151 MHz, CDCl3)δ: 142.31, 140.34, 139.80, 129.71, 129.51, 128.94, 127.97, 126.57, 51.81(CHAr2), 28.52(CH2CH3), 21.16(CH3), 15.50(CH2CH3); ESI-MSm/z: calcd for C41H46N{[M+H]+}552.3625, found 552.3618。
A2: 5.64 g,收率85%;1H NMR(600 MHz, CDCl3)δ: 7.08(d,J=7.9 Hz, 8H, Ar-H), 6.99(d,J=7.8 Hz, 8H, Ar-H), 6.39(s, 2H, Ar-H), 5.38(s, 2H, CHAr2), 3.30(s,br, 2H, NH2), 2.63~2.50(m, 8H, CH2CH2CH2CH3), 2.03(s, 3H, CH3), 1.66~1.54(m, 8H, CH2CH2CH2CH3), 1.42~1.31(m, 8H, CH2CH2CH2CH3), 0.93(t,J=7.4 Hz, 12H, CH2CH2CH2CH3);13C NMR(151 MHz, CDCl3)δ: 141.00, 140.26, 139.80, 129.74, 129.42, 128.92, 128.48, 126.52, 51.78(CHAr2), 35.35(CH2CH2CH2CH3), 33.64(CH2CH2CH2CH3), 22.54(CH2CH2CH2CH3), 21.13(CH3), 14.07(CH2CH2CH2CH3); ESI-MSm/z: calcd for C49H62N{[M+H]+}664.4877, found 664.4858。
(2) 吡啶亚胺配体的合成(Scheme 3)

Scheme 4
配体L1和L2的制备:ZnCl20.34 g(2.5 mmol)和2-乙酰吡啶2.0 mmol悬浮于冰醋酸5 mL中。再加入相应大位阻苯胺2 mmol,搅拌回流反应4 h,冷却至室温,析出亮黄色固体。过滤,滤饼用乙酸(3×5 mL)和乙醚(5×5 mL)洗涤所得固体粉末,除去残留的乙酸。在真空下干燥得到亮黄色的固体。将上一步骤的产物悬浮于二氯甲烷30 mL中,并添加草酸钾0.41 g(2.2 mmol)的水5 mL溶液。将反应混合物剧烈搅拌1 h。分离两相,用水(3×20 mL)洗涤有机层并用MgSO4干燥。过滤后,再旋蒸,除去溶剂,得到黄色粉末状产品,并在高真空下干燥。
L1: 0.92 g,收率70%;1H NMR(600 MHz, CDCl3)δ: 8.61(d,J=3.9 Hz, 1H, Ar-H), 8.10(d,J=8.1 Hz, 1H, Ar-H), 7.80~7.66(m, 1H, Ar-H), 7.42~7.30(m, 1H, Ar-H), 7.08(d,J=8.0 Hz, 4H, Ar-H), 7.02(d,J=7.9 Hz, 4H, Ar-H), 6.98(d,J=8.0 Hz, 4H, Ar-H), 6.95(d,J=8.0 Hz, 4H, Ar-H), 6.76(s, 2H, Ar-H), 5.25(s, 2H, CHAr2), 2.62(dq,J=23.1, 7.6 Hz, 8H, CH2CH3), 2.22(s, 3H, Ar-CH3), 1.24(t,J=7.6 Hz, 6H, CH2CH3), 1.21(t,J=7.6 Hz, 6H, CH2CH3), 1.13(s, 3H, Ar-C(CH3)=N);13C NMR(151 MHz, CDCl3)δ: 169.43(C=N), 156.33, 148.50, 146.10, 141.94, 141.67, 141.46, 140.19, 136.10, 132.53, 131.46, 129.79, 129.43, 128.52, 127.75, 127.51, 124.58, 121.50, 51.34(CHAr2), 28.51(CH2CH3), 28.49(CH2CH3), 21.46(Ar-CH3), 17.03(Ar-C(CH3)=N), 15.67(CH2CH3), 15.63(CH2CH3); MALDI-TOF-MSm/z: calcd for C48H50N{[M]+}654.3974, found 654.4003。
L2: 1.01 g,收率66%;1H NMR(600 MHz, CDCl3)δ: 8.59(d,J=4.1 Hz, 1H, Ar-H), 8.05(d,J=7.8 Hz, 1H, Ar-H), 7.70(t,J=7.2 Hz, 1H, Ar-H), 7.39~7.29(m, 1H, Ar-H), 7.03(d,J=7.9 Hz, 4H, Ar-H), 6.94(dt,J=11.9, 8.0 Hz, 12H, Ar-H), 6.71(s, 2H, Ar-H), 5.21(s, 2H, CHAr2), 2.65~2.46(m, 8H, CH2CH2CH2CH3), 2.19(s, 3H, Ar-CH3), 1.68~1.48(m, 8H, CH2CH2CH2CH3), 1.45~1.20(m, 8H, CH2CH2CH2CH3), 1.08(s, 3H, Ar-C(CH3)=N), 0.92(q,J=7.1 Hz, 12H, CH2CH2CH2CH3);13C NMR(151 MHz, CDCl3)δ: 169.39(C=N), 156.32, 148.49, 146.12, 141.41, 140.56, 140.32, 140.14, 136.03, 132.56, 131.40, 129.72, 129.35, 128.50, 128.30, 128.04, 124.53, 121.47, 51.35(CHAr2), 35.34(CH2CH2CH2CH3), 35.27(CH2CH2CH2CH3), 33.72(CH2CH2CH2CH3), 33.70(CH2CH2CH2CH3), 22.52(CH2CH2CH2CH3), 22.41(CH2CH2CH2CH3), 21.45(Ar-CH3), 16.99(Ar-C(CH3)=N), 14.09(CH2CH2CH2CH3), 14.05(CH2CH2CH2CH3); MALDI-TOF-MSm/z: calcd for C55H66N2{[M]+}766.5226, found 766.5245。
(3) 吡啶亚胺钯配合物的合成
在室温下,将配体L1或L20.5 mmol和催化剂前体(COD)PdMeCl(COD=1,4-环辛二烯133 mg,0.5 mmol)在CH2Cl2(10 mL)溶剂中搅拌24 h。反应过程中,溶液颜色加深。在反应结束时,溶剂在减压下部分蒸发。剩余混合物用乙醚20 mL稀释。通过过滤收集得到的黄色固体,真空干燥得Pd1或Pd2(Chart 2)。
Pd1: 0.36 g,收率89%, a-isomer/b-isomer=20/1;1H NMR(600 MHz, CDCl3)δ: 9.61, 9.34(d,J=4.3 Hz, 1H, Ar-H), 8.03, 7.90(td,J=7.8, 1.5 Hz, 1H, Ar-H), 7.75~7.77, 7.72~7.67(m, 1H, Ar-H), 7.20~7.25, 7.07~6.97(m, 8H, Ar-H), 6.84~6.96(m, 9H, Ar-H), 6.82, 6.71(s, 2H, Ar-H), 6.01, 5.85(s, 2H, CHAr2), 2.59(q,J=7.5 Hz, 4H, CH2CH3), 2.51(q,J=7.6 Hz, 4H, CH2CH3), 2.22, 2.15(s, 3H, Ar-CH3), 1.19(t,J=7.6 Hz, 6H, CH2CH3), 1.13~1.10(m, 6H, CH2CH3), 0.94(s, 3H, Ar-C(CH3)=N), -0.14, -0.15(s, 3H, Pd-CH3);13C NMR(151 MHz, CDCl3)δ: 178.19(C=N), 152.69, 149.36, 142.63, 142.25, 141.07, 140.01, 138.74, 138.36, 135.95, 135.91, 130.12, 129.39, 129.02, 128.14, 128.01, 127.64, 123.82, 51.26(CHAr2), 28.43(CH2CH3), 21.57(Ar-CH3), 17.50(Ar-C(CH3)=N), 15.96(CH2CH3), 15.54(CH2CH3), 2.57(Pd-CH3); Anal. Calcd for C49H53N2PdCl: C 72.49, H 6.58, N 3.45, found C 72.36, H 6.71, N 3.28; MALDI-TOF-MSm/z: calcd for C48H50N2Pd{[M-Cl-CH3]+}760.3009, found 760.2966。
Pd2: 0.33 g,收率70%, a-isomer/b-isomer=10/1;1H NMR(600 MHz, CDCl3)δ: 9.64, 9.35(d,J=4.4 Hz, 1H, Ar-H), 8.02, 7.88(t,J=7.2 Hz, 1H, Ar-H), 7.77, 7.69(t,J=6.7 Hz, 1H, Ar-H), 7.26~7.17, 7.04~6.91(m, 8H, Ar-H), 6.96~6.77(m, 11H, Ar-H), 6.08, 5.85(s, 2H, CHAr2), 2.55(t,J=7.6 Hz, 4H, CH2CH2CH2CH3), 2.47(t,J=7.5 Hz, 4H, CH2CH2CH2CH3), 2.22, 2.16(s, 3H, Ar-CH3), 1.59~1.53(m, 4H, CH2CH2CH2CH3), 1.49~1.43(m, 4H, CH2CH2CH2CH3), 1.33~1.25(m, 8H, CH2CH2CH2CH3), 0.94(s, 3H, Ar-C(CH3)=N), 0.91~0.84(m, 12H, CH2CH2CH2CH3), -0.15, -0.17(s, 3H, Pd-CH3);13C NMR(151 MHz, CDCl3)δ: 178.04(C=N), 152.61, 149.34, 141.07, 140.99, 140.85, 139.87, 138.65, 138.16, 135.89, 130.03, 129.22, 128.95, 128.53, 128.13, 123.62, 51.19(CHAr2), 35.15(CH2CH2CH2CH3), 34.98(CH2CH2CH2CH3), 33.77(CH2CH2CH2CH3), 33.51(CH2CH2CH2CH3), 22.32(CH2CH2CH2CH3), 22.12(CH2CH2CH2CH3), 21.49(Ar-CH3), 17.38(Ar-C(CH3)=N), 13.93(CH2CH2CH2CH3), 13.86(CH2CH2CH2CH3), 2.54(Pd-CH3); Anal. Calcd for C57H69N2PdCl: C 74.09, H 7.53, N 3.03, found C 73.86, H 7.45, N 3.13; MALDI-TOF-MSm/z: calcd for C56H66N2Pd{[M-Cl-CH3]+}872.4261, found 872.4246。
1.3 乙烯或丙烯齐聚以及其与丙烯酸甲酯共齐聚
(1) 吡啶亚胺钯配合物作为催化剂进行乙烯(丙烯)均聚的一般方法
首先在真空90 ℃下干燥一台连接高压气体管路的350 mL厚壁耐压玻璃釜,干燥时间至少为1小时;然后将玻璃釜调整到所需的聚合温度。在氮气气氛下向反应器中加入38 mL二氯甲烷和所需量的NaBArF,然后通过注射器将含所需量的吡啶亚胺钯催化剂的2 mL二氯甲烷溶液注入聚合体系中。在快速搅拌下,将反应器加压并将乙烯压力(丙烯)维持在4 atm并保持3 h后,将压力放空,在减压的条件下将二氯甲烷蒸发去除即可得到油状乙烯和丙烯的齐聚物。
(2) 吡啶亚胺钯配合物作为催化剂进行丙烯酸甲酯与乙烯(丙烯)共聚的一般方法
首先在真空90℃下干燥一台连接高压气体管路的350 mL厚壁耐压玻璃釜,干燥时间至少为1 h;然后将玻璃釜调整到所需的聚合温度。在氮气气氛下向反应器中加入二氯甲烷与所需量的极性单体的混合溶液(共18 mL)以及所需量的NaBArF,然后通过注射器将2 mL含所需量的吡啶亚胺钯催化剂的二氯甲烷溶液注入聚合体系中。在快速搅拌中,将玻璃釜加压并维持所需的乙烯(丙烯)压力并保持12 h后,将压力放空,在减压的条件下将二氯甲烷和丙烯酸甲酯蒸发去除即可得到油状共聚物。
2 结果与讨论
2.1 吡啶亚胺配体及相应钯配合物的合成
在Lucas试剂(盐酸氯化锌溶液)存在下,以对甲苯胺和二芳基甲醇为原料,进行付克烷基化反应合成大位阻的芳胺A1~A2。随后进一步采用经典的模板法[33-35]制备相应的锌配合物Zn1~Zn2, 原位用草酸钾脱除氯化锌得到相应的配体L1~L2。同时配体与(COD)PdMeCl(COD=1,5-环辛二烯)反应容易高产率(70%~89%)的得到相应的钯配合物Pd1~Pd2。这些钯配合物采用了核磁共振、质谱和元素分析进行了鉴定。1H NMR和13C NMR的分析表明,由于配体的不对称性,所获得的钯配合物是两种不同比例的顺反异构体的混合物。
2.2 吡啶亚胺钯催化的乙烯和丙烯齐聚
这些钯配合物在过量的四(3,5-二(三氟甲基)苯基)硼酸钠(NaBArF)活化下催化乙烯或丙烯齐聚(Table 1)。总体上这些钯配合物展现出较高的齐聚活性(高于104g mol-1h-1),生成低分子量高度支化的乙烯或丙烯齐聚物。这些齐聚物的分子量低于常规GPC的检出限(500 g/mol),因此所有低聚物的分子量由核磁共振氢谱根据不饱和端基的强度与总积分比确定。这些齐聚物常态下为低粘度液体。一般而言,随着聚合温度升高,这些钯配合物的活性得到进一步增强(Table 1, Figure 1a)。而分子量则随着聚合温度增加而降低(Table 1, Figure 1b)。不同的是,乙烯齐聚物的支化度随着温度升高而升高而丙烯齐聚物则基本保持不变(Table 1, Figure 1c)。这主要可能是因为乙烯齐聚物的支化度源自催化剂的链行走形成的支链而丙烯齐聚物的支化则主要归因于单体自身引入的支链。比较乙烯与丙烯齐聚,乙烯的齐聚活性要高于相应的丙烯齐聚,所得乙烯齐聚物分子量要高于丙烯齐聚物,而相应乙烯齐聚物的支化度则低于丙烯齐聚物。上述现象产生的主要原因是乙烯单体体积要小于丙烯单体,有利于乙烯的配位和插入,同时丙烯单体能自动引入支链。

图1 (a) 由Pd1~Pd2在30 ℃(前)和50 ℃(后)(表格1)条件下获得的乙烯和丙烯齐聚物的产率;(b) 由Pd1~Pd2在30 ℃(前)和50 ℃(后)(表格1)条件下获得的乙烯和丙烯齐聚物的分子量;(c) 由Pd1~Pd2在30 ℃(前)和50 ℃(后)(表格1)条件下获得的乙烯和丙烯齐聚物的支化度
进一步的核磁共振氢谱和碳谱分析表明这些齐聚物都是高度支化甚至超支化的齐聚物。我们选择表1条目2中的乙烯齐聚物样品作为示例来说明氢谱和碳谱的分析(Figure 2)。氢谱分析表明所获得的乙烯齐聚物具有高度支化,且以内双键为主要端基(Figure 2a)。此外,碳谱也进一步证实了其具有高度支化的微观结构,观察到其拥有甲基,乙基,正丙基,长链支链(>C3支链)和仲丁基支链(支链上支链的结构)等碳谱的信号峰(Figure 2b)[7]。超支化乙烯齐聚物的形成也证实了这些钯配合物对于低聚乙烯具有深度异构化的能力和表现出具有强链行走能力的催化剂特征。因此,在该体系中得到的产物可视为超支化乙烯齐聚物[7,36-37]。

表1 利用钯催化剂催化的乙烯或丙烯齐聚
2.3 吡啶亚胺钯催化的乙烯或丙烯与丙烯酸甲酯共聚
二亚胺钯和吡啶亚胺钯催化剂都能有效催化乙烯或丙烯与极性单体共聚[29-32,38-41]。本工作中,上述吡啶亚胺钯配合物在NaBArF的活化下催化乙烯或丙烯与丙烯酸甲酯(MA)共齐聚。这些配合物在乙烯与MA的共齐聚中表现出低至中等的聚合活性,得到中等至高MA插入比的低分子量高支化极性共齐聚物(Figure 3a~3d)。随着MA单体浓度的增加,聚合活性有着显著的降低而共齐聚物分子量以及支化度则保持小幅度上升,特别需要指出的是MA的插入比得到大幅度提高。这主要是由于随着极性单体浓度的增加(从1M增加到2 M),极性官能团对阳离子钯金属中心的毒化作用也随之增强和极性单体配位以及插入的概率也随之大幅度增加[29-32]。在丙烯与MA的共齐聚中,这些配合物表现出非常低的共齐聚活性,生成的丙烯-MA寡聚物拥有非常高的支化度和低的分子量。这主要是由于丙烯单体位阻较大,配位和插入能垒较高,同时这些丙烯单体自带支链。值得注意的是这些丙烯-MA寡聚物拥有非常高的极性单体插入比(24.3 mol%, Figure 3c)。这可能还是因为丙烯单体位阻较大,与MA相比,单体竞聚率相差不大所致。更进一步的,与乙烯和MA共齐聚相比,丙烯与MA共齐聚表现出非常低的共齐聚活性,相似的齐聚物分子量以及非常高的MA插入率和齐聚物支化度。上述现象表明乙烯比丙烯更易于发生配位插入,与MA相比,竞聚能力更强。此外,催化剂取代基(乙基与丁基比较)的变化就聚合活性,所得共齐聚物微结构,分子量和插入比而言对共齐聚过程影响较弱。

图2 表1中的超支化寡聚物的核磁氢谱(a)和碳谱(b)详细分析
另外,进一步利用核磁共振氢谱和碳谱分析这些所得共齐聚物的微结构。选用表2条目2中的乙烯-丙烯酸甲酯共齐聚物样品作为示例来说明氢谱和碳谱的分析(Figure 4)。氢谱表明所得共齐聚物是高度支化且端基以内双键为主(Figure 4a)。碳谱则进一步显示这些共齐聚物含有甲基、乙基、正丙基、长支链(>C3支)和仲丁基支链(支化上支化的结构)以及酯基官能团化的支链(Figure 4b)。这些结果明确地验证了所得乙烯-丙烯酸甲酯共齐聚物的超支化结构,且酯基位于支链的末端,与先前报道的α-二亚胺钯(II)催化剂和吡啶亚胺钯(II)催化剂相一致[29-32,38-41]。本体系中获得的超支化极性官能团化低聚物可能在粘度改性剂或界面活性剂等领域中有潜在应用价值[5-6]。
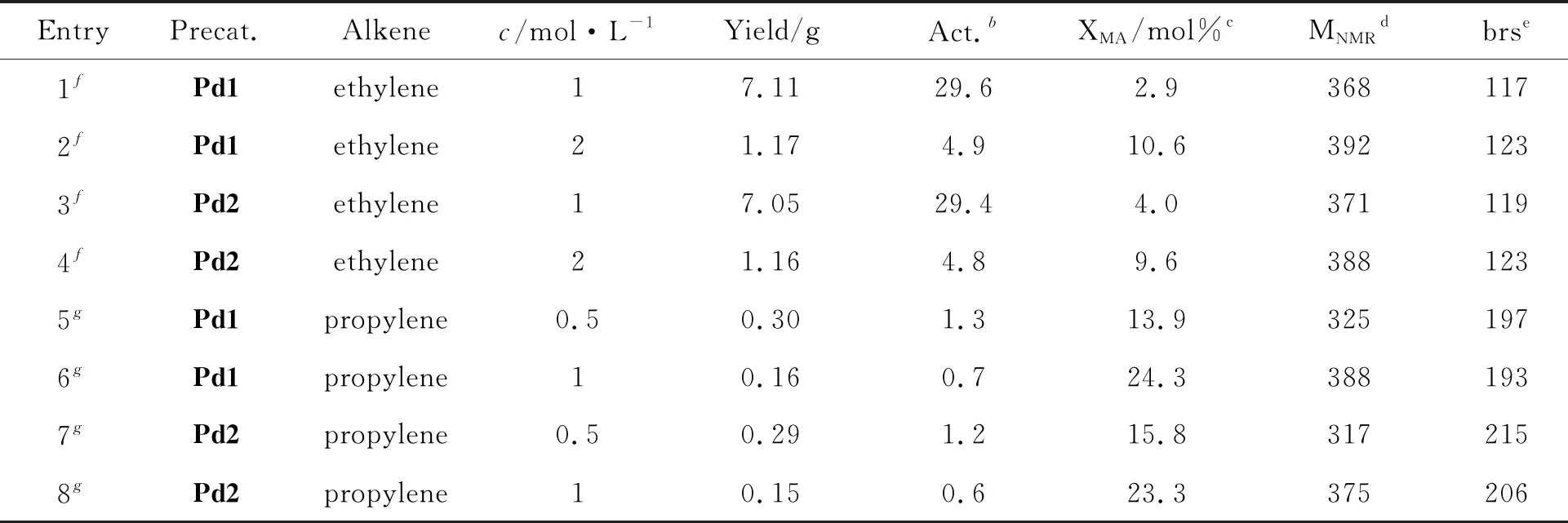
表2 利用钯催化剂催化的乙烯或丙烯与丙烯酸甲酯共齐聚

图3 (a) 由Pd1~Pd2在1 M(前)和2 M(后)(表格2)条件下获得的乙烯-丙烯酸甲酯和丙烯-丙烯酸甲酯共齐聚物的产率;(b) 由Pd1~Pd2在1 M(前)和2 M(后)(表格2)条件下获得的乙烯-丙烯酸甲酯和丙烯-丙烯酸甲酯共齐聚物的分子量;(c) 由Pd1~Pd2在1 M(前)和2 M(后)(表格2)条件下获得的乙烯-丙烯酸甲酯和丙烯-丙烯酸甲酯共齐聚物的支化密度

图4 表2,条目2的超支化共寡聚物的核磁氢谱(a)和碳谱(b)详细分析
合成了一类新型吡啶亚胺钯催化剂,研究了其用于乙烯和丙烯齐聚及乙烯或丙烯和极性单体MA共聚。在乙烯齐聚和丙烯齐聚中,该催化剂均表现出中等齐聚活性,可分别催化得到超支化(112~130/1000C)的低分子量(300~500 g/mol)乙烯齐聚物和超高支化(235~238/1000C)的低分子量(约200~300 g/mol)丙烯齐聚物。进一步,将该钯催化剂用于乙烯或丙烯和MA共齐聚。前者表现出低至中等的共齐聚活性(103~104g mol-1h-1),可生成超支化(117~123/1000C)的低分子量(300~400 g/mol)乙烯-MA共齐聚物,且这些共齐聚物拥有非常高的极性单体插入比(高达10.6 mol%)。后者表现出高效的共齐聚特性,可催化得到高度支化(193~215/1000C)高插入比(高达24.3 mol%)的低分子量(约300~400 g/mol)丙烯-MA共齐聚物。以上结果表明该催化体系不仅能有效催化乙烯和丙烯齐聚制备超支化甚至超高支化的乙烯及丙烯齐聚物,还能催化乙烯或丙烯与MA共齐聚制备相应极性官能团化的超支化共齐聚物。
猜你喜欢
分析测试学报(2022年10期)2022-10-22
检验医学(2022年2期)2022-03-14
合成化学(2022年1期)2022-02-19
农业机械学报(2021年10期)2021-11-09
中华养生保健(2020年3期)2020-11-16
今日农业(2019年11期)2019-08-13
成长·读写月刊(2017年3期)2017-04-08
科技视界(2016年26期)2016-12-17
北京航空航天大学学报(2014年1期)2014-12-19
食品工业科技(2014年13期)2014-12-16
