
用于甲醇重整制氢的铜基催化剂研究进展
2022-01-10 03:08孙晓明沙琪昊王陈伟周道金
化工学报 2021年12期
孙晓明,沙琪昊,王陈伟,周道金
(北京化工大学化学学院,化工资源有效利用国家重点实验室,北京 100029)
引 言
近年来,伴随着世界各国的快速发展,温室气体被大量排放。二氧化碳的排放速率远超过其被陆地和海洋吸收的速率,其带来的温室效应以及极端厄尔尼诺现象严重影响了人类的生活质量[1],并造成了极大的经济与生态损失。为了减少二氧化碳的排放量,以便将全球温度稳定在国际社会力图达到的目标水平[2-3],世界各国纷纷开展各自的“碳中和”计划[4]。氢气因其热值高、无污染等特点,被学者誉为21世纪的终极能源[5],但要在未来能源基础设施中广泛使用氢,进入“氢经济(hydrogen economy)”时代,必须应对许多关键挑战(如氢气的制备、储存、运输等)[6]。
在氢气的制备方面,目前工业上应用广泛的煤制氢和天然气制氢虽然技术成熟,但这种氢气的产生以化石能源的消耗为代价,并不满足可持续发展的要求;电解水制氢技术虽然设备简单,氢气纯度高,但其成本过高,多用于小型制氢;在众多制氢方式当中,甲醇制氢日益受到科研工作者的青睐,以甲醇水蒸气重整为代表的甲醇制氢技术具有较低的反应温度,产生氢气纯度较高,投资少,能耗低,且原料甲醇运输存储方便,便于进行分布式生产[7-8]。
利用可再生能源,将二氧化碳和水制备成“液态阳光甲醇”(图1)[9-10],可以逐步淘汰煤、石油等化石燃料[11-12];继续利用甲醇作为储氢载体[13-14],水蒸气重整反应产生氢气,整个过程安全可靠[15-16]。将甲醇重整制氢反应与工业加氢反应,如合成氨、费-托合成、乙炔加氢反应等耦联,可以实现一大批高值化学品如化肥、液态烃或碳氢化合物及高纯度乙烯的制备。因此大力发展高效甲醇重整制氢对于化工领域的高纯度、高利润发展有重大意义。

图1 (a)液态阳光愿景[9];(b)液态阳光路线图[9];(c)利用可再生能源制备甲醇,甲醇作为能量载体用于运输和存储,以及在终点释放氢气[10];(d)通过甲醇收集可再生能源的波动的能量,可使用相同的催化剂及转化器来驱动甲醇合成和甲醇水蒸气重整[10]Fig.1 (a)Liquid sunshine vision[9];(b)Liquid sunshine roadmap[9];(c)Renewable energy is used to produce methanol,which is used as an energy carrier for transportation and storage,and releases hydrogen at the end[10];(d)The fluctuating energy of renewable energy is collected by methanol,and the same catalyst and converter can be used to drive methanol synthesis and methanol steam reforming[10]
目前甲醇重整制氢方法主要有四种,分别是:甲醇分解(MD)[式(1)]、甲醇部分氧化重整(POM)[式(2)]、甲醇水蒸气重整(MSR)[式(3)]、氧化甲醇蒸汽重整(OSRM)[式(4)][17]。

在四种制氢方法中,甲醇水蒸气重整制氢是研究得最广泛、最深入的制氢方法。相较于其他制氢方式,甲醇水蒸气重整制氢的优点在于反应条件温和,且具有较低的CO含量,该制氢方式每摩尔甲醇可以产生的氢气量最高[18]。目前应用于甲醇水蒸气重整制氢的催化剂主要有两类:一类是贵金属催化剂(如Pd/ZnO等);另一类是非贵金属催化剂,包括非铜基催化剂(如Zn-Cr等)和铜基催化剂(如CuO/ZnO/Al2O3等)。
贵金属催化剂的优点是有较高的稳定性,但活性和选择性不如Cu基催化剂,且气体产物中CO含量较高。Iwasa等[19]研究了负载在SiO2载体上的Cu基催化剂和几种贵金属催化剂(Rh、Pt、Pd)的MSR性能,结果表明Cu/SiO2具有最好的MSR性能,氢气产率为83.4 μmol·(g cat)-1·min-1,远超其他贵金属催化剂;在CO选择 性方面,Cu/SiO2的CO选 择性仅为6%,而其他贵金属催化剂CO选择性均为100%,这表明在贵金属催化剂上甲醇全部分解变成CO和H2。Iwasa等认为铜和贵金属催化剂催化性能的差异归因于反应过程中形成的HCHO物种反应活性不同。
随后Iwasa等[20-21]将Pd、Pt负载在不同载体上,探究载体类型不同对反应性能的影响。结果表明,Pd/ZnO催化剂具有最好的催化性能。相比于Pd/SiO2催化剂100%的CO选择性,Pd/ZnO催化剂的CO选择性仅为0.8%。Iwasa等[22]进一步对Pd/ZnO催化剂进行探究,通过不同还原温度预处理下的X射线衍射光谱(XRD)发现[图2(b)],随着还原温度的升高,Pd逐渐与Zn形成PdZn合金,Iwasa等指出Pd/ZnO对CO的低选择性归因于PdZn合金的生成。

图2 (a)Zn-Cr催化剂甲醇蒸汽重整转化率和产物分布与反应温度的关系[24];(b)不同预还原温度下Pd/ZnO的XRD谱图[22]Fig.2 (a)Methanol conversion and product distribution for steam reforming vs reaction temperature based on Zn-Cr catalyst[24];(b)XRD patterns of Pd/ZnO with different reduction pretreatment temperatures[22]
负载在In2O3、Ga2O3上的Pt、Pd催化剂同样表现出了较高的蒸气重整选择性和较低的CO选择性,Iwasa等[20-21]对这些催化剂进行了X射线衍射(XRD)和X射线光电子能谱(XPS)表征,证实了Pd-In、Pd-Ga、Pt-Zn、Pt-In、Pt-Ga合金相的形成,而负载在ZnO上的Co、Ni、Ru、Ir催化剂并没有形成合金相,展现出较差的MSR选择性。
表1对比了相同温度下多种贵金属催化剂MSR性能。可以看到Pd/ZnO催化剂在选择性方面具有明显的优势。但Pd的高昂价格限制了其在实际大规模工业化的应用,因此研究希望得到廉价而又高效的催化剂,后续的研究继续发现,非贵金属催化剂如非铜基催化剂(如Zn-Cr等)和铜基催化剂(如CuO/ZnO/Al2O3等)也被广泛应用于甲醇水蒸气重整制氢体系。

表1 不同载体的贵金属催化剂金属结构及MSR选择性Table1 Metal structure and MSR selectivity of precious metal catalysts on different supports
以Zn-Cr催化剂为代表的非铜基催化剂相较于Cu基催化剂有较好的热稳定性[23],且在高温时有与Cu基相似的催化性能,但其缺点是在低温时反应活性差,且产物中CO含量较高。Cao等[24]采用共浸渍法制备了以γ-Al2O3为载体的Zn-Cr催化剂,并在H/C=1.4 ,GHSV=25000h-1条 件下 测 试MSR性能,图2(a)为反应温度对Zn-Cr催化剂上甲醇转化率和产物分布的影响,在380~460℃范围内H2和CO2的比例约为3∶1,CO是主要的副产物。相比于Cu基催化剂,Zn-Cr催化剂需要一个更高的反应温度才能达到100%甲醇转化率,在GHSV=25000h-1下反应温度须达到460℃,此时产物中CO含量为1.5%。因为Zn-Cr基催化剂仅在高温时才有较好的催化表现,在低温时的甲醇转化率和CO选择性均不如Cu基催化剂,而在实际应用中,反应常常在较低温度范围内进行,所以本文仅在机理部分对Zn-Cr催化剂CO产生原因及调控方法进行简单介绍,其余部分不涉及Zn-Cr基催化剂。
Cu为成本低廉的非贵金属,相较于贵金属催化剂和以Zn-Cr为代表的非Cu基催化剂,Cu基催化剂可在低温下高选择性地生成CO2和H2,同时具有较低的CO选择性,因此Cu系催化剂被广泛应用[25]。研究者对Cu基催化剂进行了大量研究,包括掺入不同的助剂、使用不同的载体、构造不同的结构等,均取得了良好的研究成果(表2)。本文首先讨论了目前甲醇水蒸气重整的机理,然后讨论了Cu活性价态和活性晶面,并基于机理讨论总结了高效催化剂的设计进展,包括复合氧化物与铜的界面效应以及氧空位的影响,接着探讨了催化剂失活机制,最后对未来甲醇水蒸气重整催化剂的发展进行了展望。

表2 贵金属催化剂、Zn-Cr催化剂与部分Cu基催化剂MSR催化效能汇总Table2 Summary of MSR catalytic performance of precious metals,Zn-Cr and some Cu-based catalysts
1 反应机理
甲醇水蒸气重整(MSR)在铜基催化剂上的反应机理到目前为止仍未有统一的结论。起初研究者们认为甲醇通过分解生成氢气和一氧化碳[式(5)],然后再进行水汽反应将部分一氧化碳转变为二氧化碳,同时生成氢气[式(6)][29-30]:
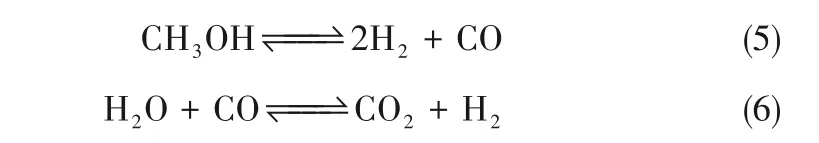
根据该机理,产物中的一氧化碳浓度应该会高于该温度下的热力学平衡浓度[31]。但研究结果表明铜基催化剂产生的一氧化碳浓度低于平衡时的浓度[29,32],因此Takahashi等[33]否定了这种反应顺序,并提出了一种涉及甲酸甲酯中间体的机理[式(7)~式(9)]:

在后续工作中,Jiang等[34]也支持这个机理。甲醇脱氢生成甲酸甲酯和氢气之后,甲酸甲酯被水解形成甲酸和甲醇,然后甲酸分解形成二氧化碳和氢气[35]。由于CO不是通过甲醇分解而形成的,因此认为其来源于逆水汽反应(reverse water gas shift,RWGS)[式(10)]:

在活性位点方面,Jiang等仅描述了一种类型的活性部位,而Peppley等[36]提出了两种类型的活性位点:一种位点对MSR和水煤气变换反应具有活性,而另一种位点主要支持分解反应。此外,这些研究假设氢吸附并不会与含氧物种吸附的活性位点竞争。
Iwasa等[19]在铜基催化剂的蒸汽重整反应中发现了甲醛和甲酸的中间体,因此提出了另一种机制[式(11)~式(13)]:首先甲醇脱氢生成甲醛中间体,随后甲醛中间体被水进攻生成甲酸,最后甲酸分解生成二氧化碳和氢气。Breen等[37]和Shishido等[38]也证实了这种机制,并提出CO的形成可能是由于逆水汽反应。

Frank等[39]提出了一个完整的MSR催化循环,汇编了Jiang等、Peppley等和Iwasa等提出的几种机制。如图3(a)所示,催化循环始于甲醇在催化剂表面的解离吸附,并假设活性位点A负责含氧化合物的吸附,且以单层竞争性形式吸附;活性位点B负责氢的吸附,氢在活性位点上以游离形式存在。在此循环中描述了Zhang等[40]提出的机制:甲醇脱氢形成的甲醛受到甲氧基攻击后,生成中间体甲酸甲酯,后者可以与羟基反应分解为甲氧基和甲酸基。该循环中描述的另一种机理是:羟基和甲醛反应形成二氧亚甲基,随后二氧亚甲基脱氢成甲酸基,甲酸基再进一步脱氢产生二氧化碳和氢气。
Lin等[41]、Papavasiliou等[42]、Gu等[43]的 研 究 表 明甲醇脱氢生成的CH2O*可与水离解生成的OH*或O*发生偶联反应,生成羟基甲氧基(H2COOH*)或二氧亚甲基(H2COO*)中间体。中间产物依次脱氢生成CO2,而H2COOH*脱 氢 存 在C—H键 断 裂(HCOOH*)和O—H键(H2COO*)断裂的差异。Wang等[44]认为CH2O*和OH*/O*反应时,会出现三种竞争途径,包括H2COOH*/H2COO*中间体形成、CH2O*脱氢生成CO*和CH2O*解吸[图3(b)]。不同的反应路径会造成不同的CO2、HCOOH、CO和CH2O选择性,与解吸和耦合过程相比,在所有Cu晶面上,CH2O*脱氢成CO*反应具有最高的能垒(图4),而在图3(b)中可以发现,这是仅有的一条生成CO的路径,这表明Cu基催化剂上MSR的CO选择性应该非常低。

图3 (a)基于Jiang等、Peppley等、Iwasa等研究的甲醇水蒸气重整催化循环,包括不同种类的反应性表面位点A()和B()[39];(b)经由CH2O中间体的MSR反应网络[44];(c)还原的CuO-CeO2在MSR反应中的原位红外谱图[45];(d)还原的CuO-CeO2-I在MSR反应中的原位红外谱图[45]Fig.3 (a)Catalysis cycle of methanol steam reforming on the basis of the investigations of Jiang et al,Peppley et al and Iwasa et al,including different kinds of reactive surface sites A()and B()[39];(b)Reaction network of MSR via CH2O intermediate[44];(c)DRIFT spectra for MSR on reduced CuO-CeO2[45];(d)DRIFT spectra for MSR on reduced CuO-CeO2-I[45]

图4 Cu(111)、Cu(100)、Cu(110)、Cu(211)和Cu(221)上甲醛反应的能量分布。反应从共同的能量参考CH2O*+O*+OH*开始,并考虑了四种竞争反应途径:(1)CH2O*解吸(蓝色);(2)CH2O*脱氢生成CO*,然后CO*与O*反应生成CO2*(红色);(3)CH2O*与O*反应生成H2COO*,然后脱氢生成HCOO*(绿色);(4)CH2O*与OH*反应生成H2COOH*,然后脱氢生成CO2*(黑色)[44]Fig.4 Energy profiles for the formaldehyde reaction on Cu(111),Cu(100),Cu(110),Cu(211),and Cu(221).Reaction starts from a common energy reference CH2O*+O*+OH*,and four competitive reaction pathways are considered:(1)CH2O*desorption(blue);(2)CH2O*dehydrogenation to CO*followed by CO*reaction with O*to CO2*(red);(3)CH2O*reaction with O*to form H2COO*followed by its dehydrogenation to HCOO*(green);(4)CH2O*reaction with OH*to form H2COOH*followed by its dehydro-genation to CO2*(black)[44]
对于MSR反应速控步的研究,Frank等[39]利用Peppley等提出的甲醇水蒸气重整反应速率方程式[式(14)],再通过漫反射傅里叶变换红外光谱(DRIFTS)识别表面物种和中间体,得出结论:对于MSR,CH3O*脱氢生成CH2O*为整个反应的速控步骤[rate determining step(RDS),图3(a)、(b)红色剪头所指的反应步骤]。

Chen等[45]结合前人工作并结合原位红外谱图数据[图3(c)、(d)]提出了新的机理观点:CH3OH优先吸附在铜颗粒上形成甲氧基,H2O吸附在氧化铈的氧空位上,作为氧源。氧物种通过反向溢出迁移到铜表面,与甲氧基反应生成甲酸盐。甲酸盐直接分解为一氧化碳,或进一步氧化为碳酸盐,再生成二氧化碳。
尽管Cu基催化剂MSR机理存在争议,但反应步骤大致有如下部分:甲醇脱氢生成CH3O*,CH3O*脱氢生成CH2O*,水离解生成OH*或O*,而后氧物种与CH2O*进行偶联反应生成中间体,中间体进一步脱氢形成二氧化碳或一氧化碳。其中CH3O*脱氢生成CH2O*为整个反应的速控步骤[26]。对于反应中的活性位点,认为含氧中间体和氢原子分别吸附于不同的反应位点,含氧中间体以单层竞争性吸附形式存在,而氢原子以游离形式存在。对于氧物种与CH2O*偶联发生的竞争反应,CH2O*脱氢成CO*的反应具有最高的能垒,这可能是造成Cu基催化剂具有较低CO选择性的重要原因。
Iwasa等[19]提出了贵金属催化剂(Rh、Ru、Pt、Pd)的反应机理[式(15)],并认为贵金属(Rh、Ru、Pt、Pd)和铜基催化剂催化性能的差异是由于反应过程中形成的HCHO物种吸附结构不同。对于Cu、Ag等IB金属,HCHO以η1(O)-HCHO结构吸附[19,46],该结构保留了HCHO的分子特性,使得HCHO易受到亲核试剂如CH3OH(CH3O-)和H2O(HO-)的攻击,最终生成CO2、H2、HCOOCH3。而 对 于 贵 金 属,中 间 产 物HCHO以η2(C,O)-HCHO结构吸附[47-49],由于金属中的电子易返回到醛的反键轨道中,导致该结构中C—C和C—H键容易迅速断裂,生成CO和H2,因此贵金属催化剂在MSR反应中主要生成CO和H2。

随后,Iwasa等[20-21]发现在ZnO、In2O3、Ga2O3的存在下,Pd和Pt的MSR催化性能也得到了极大的改善,这归因于Pd-In、Pd-Ga、Pd-Zn、Pt-In、Pt-Ga、Pt-Zn合金相的形成。Iwasa等[20-21]指出造成合金相和金属相催化性能差异的原因也是由于反应过程中形成HCHO中间体吸附结构的差异。在Pd、Pt合金上,HCHO被稳定下来,像在铜基催化剂上一样转化为CO2、H2,而在纯金属催化剂上,HCHO极易直接分解形成CO和H2,因此合金相的贵金属催化剂相较于金属相具有更低的CO选择性。
图5显示了金属基催化剂(Rh、Ru、Pt、Pd)和Pd、Pt合金催化剂上的MSR反应路径[20]。在Pd、Pt合金上形成的甲醛中间体具有和Cu基催化剂相同的吸附结构,容易被水进攻,生成为CO2和H2。相比之下,在金属基催化剂(Rh、Ru、Pt、Pd)上的甲醛以η2(C,O)-HCHO结构吸附,甲醛易分解为CO和H2。

图5 (a)甲醛在Cu、Pt合金、Pd合金上的η1(O)-HCHO吸附结构[19];(b)甲醛在金属Rh、Ru、Pt、Pd上的η2(C,O)-HCHO吸附结构[19];(c)甲醇在Pt、Pb合金和金属Rh、Ru、Pt、Pd上的水蒸气重整反应机理[20]Fig.5 (a)The η1(O)-HCHO adsorption structure of formaldehyde on Cu,Pt alloy and Pd alloy[19];(b)The η2(C,O)-HCHO adsorption structure of formaldehyde on metal Rh,Ru,Pt,Pd[19];(c)Reaction mechanisms for the steam reforming of methanol over Pd,Pt alloys and metallic Rh,Ru,Pt,Pd[20]
关于Cu基和Pd基等贵金属反应机理已经有大量研究,但Zn-Cr催化剂上反应机理却仍不清楚。为了抑制Zn-Cr催化剂中CO生成,Cao等[24]对Zn-Cr上CO生成途径进行了研究,结果显示CO主要通过甲醇分解(MD)反应生成,CO2和H2主要通过MSR反应生成。水煤汽反应(WGS)和逆水煤汽反应(RWGS)对CO生成的影响都可以忽略。Cao等进一步探究了进料组成对产物的影响,在进料中添加H2和CO不会影响MSR反应的过程,添加CO2和H2O会降低CO2的产量。其中添加H2O对MSR反应影响最大,随着水用量的增加,CO2和CO产量均下降,但CO下降速率大于CO2,是否可以通过调控水醇比,在适当的范围内控制CO产量,以降低Zn-Cr催化剂的CO选择性,是一个值得讨论的问题(图6)。

图6 H2、CO2、CO、H2O在410℃(空心符号)和450℃(实心符号)下对MSR反应的影响[24]Fig.6 Effect of H2,CO2,CO and H2O on the MSR reaction at410℃(open symbol)and450℃(solid symbol)[24]□■CO2;△▲CO;○●H2(GHSV=2500h-1)
通过上述研究可知,贵金属催化剂和Zn-Cr催化剂在MSR中,均有极大的可能性进行生成CO的副反应,然而Cu基催化剂产生CO部分的路径具有最高的势垒,其副反应最不容易发生,这也从根本上保证了Cu基催化剂相较于Zn-Cr基和贵金属催化剂具有最低的CO选择性。因此利用Cu基催化剂通过甲醇水蒸气重整制备氢气,得到的目标产物纯度最高,可以最大程度上避免氢氧燃料电池中阳极CO毒化的问题[50-51]。
2 Cu的调控
Cu作为甲醇水蒸气重整催化剂中的活性金属,直接对其进行调控可以有效改善催化效能。本节从铜的活性晶面、活性物种两部分展开,解释暴露何种晶面最有利于MSR催化活性的提高;同时对目前争议较大的Cu基催化剂活性价态问题以及不同的价态对应的作用进行了讨论。
2.1 Cu的活性晶面
在对Cu催化剂表面进行MSR机理探索的过程中,研究者发现不同的晶面具有不同的反应活性。如图7所示,Wang等[44]通过速率控制程度分析(DRC)发现,在大多数平面上,MSR的活性受CH3O*脱氢生成CH2O*控制,这印证了反应机理部分Frank等[39]的结论,所以CH3O*在Cu表面的丰度尤为关键;其次,Cu表面应有足够丰富的O*/OH*物种参与和CH2O*的偶联反应,进而反应生成H2和CO2;而空位(*)的存在有助于避免表面中毒,为反应提供位点。所以,能提供CH3O*、O*、空位(*)最优分布的表面将呈现出较高的MSR活性。通过微观动力学模拟发现,Cu(110)表面的这三种物质有最为均匀的覆盖分布[图7(b)],使得Cu(110)有最高的CO2选择性。而Cu(111)表面全被O*覆盖导致中毒,(221)表面较低的O*丰度和(211)表面反应空位的缺失则导致其不适用于MSR反应。

图7 在Cu(111)、Cu(100)、Cu(221)、Cu(211)和Cu(110)上CO2形成的基本步骤的DRC与温度的关系(a)[44];CO2生成速率、表面物种的丰度随温度的变化(b)[44]Fig.7 DRC of the elementary steps for CO2formation(a)[44],and activity trend of CO2formation and the coverages of the most abundant species(b)on Cu(111),Cu(100),Cu(221),Cu(211),and Cu(110)as a function of temperature[44]
Hansen[52]利用原位透射电子显微镜发现,在不同气态环境下,由于吸附质引起的表面能变化和界面能变化,使得纳米Cu晶体经历可逆变化。催化剂表面有水分子的情况下,Cu(110)晶面暴露增多[图8(a)]。结合上文可知,Cu(110)具有优异的MSR活性,因此催化剂表面水分子的存在对MSR反应是有利的。
同时,Wang等[53]通过密度泛函理论(DFT)计算Cu(111)、CuZn合 金 和Cu/ZnO界 面 上 的MSR途 径[图8(b)、(c)]。H和Cu/ZnO界面上O的强相互作用促进了H2O、CH3OH和H2COOH中的H吸附和O—H键断裂,从而导致了催化剂具有高CO2和H2选择性。相反,在Cu和CuZn合金上弱的H-金属相互作用抑制了H的吸附和O—H键的断裂,导致了催化剂具有较低活性和较高的CH2O选择性。H结合的不同活性位点还导致H2COO*(Cu/ZnO界面)或HCOOH*(Cu和CuZn合金)中间体形成,使得产生CO2的反应途径发生变化。
综上,Cu(110)晶面对MSR反应表现出最好的活性和CO2选择性,因此在制备催化剂过程中,应尽可能多地暴露Cu(110)晶面。在真实的反应过程中,Cu基催化剂往往会发生一些价态和结构的改变。一般来说,Cu基催化剂在反应器中会先进行预还原,使得Cu的价态保持零价,而在反应过程中Cu可能会被部分氧化,因此关于Cu的活性价态的有关问题同样值得讨论。
2.2 Cu的活性价态
一直以来关于Cu基催化剂在MSR反应中的活性价态是零价的还是一价的问题争议不断。在不同的反应条件下,Cu的价态往往是动态变化的,因此二者很可能共存且存在协同作用。
Yang等[54]通过同位素标记、原位红外、密度泛函(DFT)计算得出结论:在甲醇脱氢生成甲酸甲酯反应中,零价铜位点促进了CH3OH的O—H键和CH3O的C—H键裂解,而一价铜位点则使得HCHO快速分解为CO和H2。
Das等[55]构建了催化剂表面的Cu物种变化模型[图9(a)]。未反应的催化剂由CeO2/ZrO2载体和CuO微晶构成,载体表面还有部分还原的铜颗粒(Cu0、Cu+),在MSR反应过程中,氧化铜被还原成Cu0纳米颗粒;随着反应时间的继续增加,Cu0纳米微晶逐渐团聚成大的颗粒,同时催化剂表面发生碳沉积;进行氧化再生后,焦炭气化,大的团聚铜颗粒分裂,并在催化剂表面发生重排,重排后催化剂表面的组成与新鲜样品组成类似。
Das等认为Cu0与CO2进行逆水汽反应(reverse water gas shift,RWGS)生 成CO。而Turco等[56]认为CO与Cu+的相互作用较强,在Cu+的存在下CO可以与表面氧物种反应生成CO2[式(17)],通过抑制RWGS反应,降低CO选择性。

因此,为了在MSR过程中保持较低CO选择性,需要维持一个特殊的Cu+/Cu0比例。Oguchi等[57]制备了CuO含量为80%(质量)的CuO/ZrO2催化剂,探究了在250℃条件下反应2h,不同的水醇比对催化剂中铜物种的影响。他们发现水占比过多会阻止一部分氧化铜的还原,导致较低的H2产量。随着水醇比的增加,催化剂中Cu+物种呈现先增加后减少的趋势,当水醇比为1.5 时,催化剂中Cu2O含量达到了90%,此时该CuO/ZrO2催化剂拥有最高的H2产率[图9(b)、(c)]。

图9 (a)不同条件下CeO2/ZrO2催化剂表面状态示意图[55];(b)水醇比对250℃下CuO/ZrO2催化剂性能的影响[57];(c)XANES光谱线性拟合获得的CuO/ZrO2催化剂上铜物种的组成[57]Fig.9 (a)Schematic diagram of the surface state of CeO2/ZrO2catalyst under different conditions[53];(b)Effect of water/MeOH ratio on the performance of CuO/ZrO2catalyst at250℃[55];(c)The composition of copper species on CuO/ZrO2catalyst obtained by linear fitting of XANES spectrum[55]
根据上文描述,DFT计算所得Cu(110)面具有良好的催化活性,因此选择性暴露该面至关重要。而且Cu基催化剂的高活性往往是需要Cu0、Cu+并存的。因此在设计高MSR催化活性的Cu基催化剂时,可以通过调控活性金属Cu入手,通过暴露更多的Cu(110)晶面,增加Cu在催化剂表面的分散,调控Cu的价态,来使催化剂达到更好的催化活性。
在直接对活性金属Cu的调控基础上,通过将Cu负载到不同金属氧化物基底表面,可以实现活性金属Cu的高度分散,此外利用复合金属氧化物和铜之间的协同作用、调控反应中间体在活性中心吸附强度等方式可有效调控催化剂性能。
3 复合氧化物的界面效应
基于以上分析,众多学者开展了一系列针对高性能Cu基催化剂的合成与优化。Cu/ZnO催化剂开始是为甲醇合成设计的,因其在250~300℃的低操作温度下具有较高活性,也被应用在甲醇水蒸气重整当中。在随后的发展中,不同助剂掺杂的Cu/ZnO催化剂也被不断研发出来,本部分从Cu/ZnO催化剂开始,总结了不同助剂掺杂的Cu基催化剂研究进展,并探究了制备不同复合氧化物所造成的界面效应对催化效能的影响。
3.1 Cu/ZnO界面效应
Cu/ZnO催化剂的界面效应一直被科学家们广泛讨论,其中最为著名的理论有溢出模型、形态模型和CuZn合金模型,同时还有学者指出Cu/ZnO催化剂中的活性位点是ZnOx修饰的铜台阶。本节从ZnO与Cu的相互作用出发,针对界面效应对MSR过程的影响进行阐述。
研究初期,Klier等[58-59]首先提出了Cu是在间隙和置换位点处结合到ZnO相中的,并指出缺陷结构以及催化剂的体积决定了催化活性。随后,Chinchen等[60]提出:在Cu/ZnO催化剂中只有Cu有催化活性,而ZnO只是稳定了一个更高的铜表面积,在他们看来,ZnO只是一种惰性载体,Chinchen等的结论基于观察到的催化活性与铜表面积之间的线性关系。
Burch等[61]和Spencer[62]否认了ZnO仅是一种惰性载体的说法,并提出了溢出模型理论。他们认为,Cu和ZnO之间的氢双向转移是至关重要的,由于ZnO具有较大的吸附氢容量,它可能会捕获原产生于Cu表面的氢原子,并充当储氢器,从而加速催化剂中氢的溢出。但是这种效应只有在部分氧化的Cu表面才能快速发生,在完全还原的Cu表面,氢原子的溢出会受到限制。同时Spencer[62]的研究发现吸附在Cu0上的甲酸通过ZnO的氢溢出进行氢解。Nakamura等[63]在甲醇合成中证实了溢出效应的存在,但认为与创建活性位点的作用相比,ZnO的主要作用并不是溢出效应。
Topsøe等[64]提出的形态模型认为,ZnO表面存在直径为1~1.5 nm的铜颗粒,这些铜颗粒的形态会随着催化剂氧化还原处理条件而变化,这归因于Cu-ZnO界面的润湿/非润湿现象。在部分还原的ZnO表面,铜颗粒的形状接近于圆盘,而在完全氧化的ZnO表面上,由于铜氧化锌之间的弱相互作用,铜颗粒变得更接近球形[图10(a)]。
而相比于ZnO负载Cu颗粒的形态模型,CuZn合金模型被更多科学家所认同。CuZn合金模型认为ZnO像垫片一样把Cu颗粒分隔开,ZnO作为隔离物,防止铜颗粒烧结[图10(a)]。Nakamura等[63]通过X射线衍射(XRD)表征发现了Cu的晶格常数变化,证实了40%~60%(质量)的CuZn合金的形成。Grunwaldt等[65]发现表面上的CuZn合金可能是由ZnO迁移到Cu0微晶所形成的。Topsøe等[64]对甲醇合成过程中ZnO催化剂上的铜进行了原位红外测量,根据观察到的CO能带位移,推断了ZnO部分迁移到还原铜微晶上时表面CuZn合金的形成。同时Topsøe等[64]还指出CuZn合金只有在Cu/ZnO高温还原(873K以上)后才能形成。
Nakamura等[63]在对形态模型和CuZn合金模型的讨论中指出,工业催化剂中铜和氧化锌含量通常相当,并且铜和氧化锌是以混合形式存在,而非氧化锌支撑铜颗粒,因此认为CuZn合金的形成是促进氢制甲醇活性的主要原因。Kasatkin等[66]通过透射电子显微镜(TEM)表征证实了氧化锌颗粒充当铜颗粒之间的隔离物,防止它们烧结[图10(b)]。这与CuZn合金模型相符。

图10 (a)Nakamura等[63]的CuZn合金模型,Topsøe等[64]的铜形态效应模型;(b)透射电镜和高分辨率透射电镜揭示的显微结构特征[66];(c)常规制备的活性最高的Cu/ZnO/Al2O3催化剂中铜粒子的像差校正HRTEM图像[67]Fig.10 (a)Model of Cu-Zn alloy by Nakamura et al[63];Model of Cu morphology effect by Topsøe et al[64];(b)Microstructural features revealed with TEM and high-resolution TEM[66];(c)Aberration-corrected HRTEM images of Cu particles in the conventionally prepared,most-active Cu/ZnO/Al2O3catalyst[67]
Cu/ZnO中还存在很多结构缺陷,如孪晶、堆垛层错,还有学者提出Cu/ZnO中的活性位点是被ZnOx修饰的铜台阶,这被认为加强了中间物质和催化剂之间的相互作用[65]。如Kasatkin等[66]所得电镜图所示,氧化锌颗粒充当铜颗粒之间的隔离物,通常铜颗粒与几个氧化锌颗粒接触,铜表面被氧化锌部分覆盖。大多数铜颗粒呈现类似于椭球或球体的圆形(非平衡)形状。多数铜颗粒,尤其是大于5nm的铜颗粒,含有各种缺陷,其中最典型的是孪晶和堆垛层错。
Behrens等[67]的研究发现由ZnOx修饰的Cu台阶[图10(c)]是高效Cu基催化剂的活性位点,因为中间体在台阶位点有更强的结合力和更低的结合能垒,从而导致了更高的催化性能。而台阶位点则可以通过体缺陷如堆垛层错或孪晶来稳定。同时Behrens等通过高分辨透射电镜(HRTEM)还发现铜表面存在分布不规则的ZnOx纳米粒子,Znδ+的存在增加了HCO、H2CO、H3CO在台阶状活性位点的吸附强度,减小了吸附势垒,从而进一步提高了Cu/ZnO的催化活性。
综上所述,Cu/ZnO及其衍生物催化剂作为催化剂组分相对单一、研究对象最为清晰的一类催化剂,受到了广泛关注。高分辨成像技术、多尺度原位谱学技术对其在催化过程中的结构、形貌和配位状况的演变提供了诸多直接证据,也为辨认活性位点,研究界面效应对催化活性、稳定性的影响提供了依据。目前研究通过实验结果推论以Cu/ZnO为前物,在反应条件下形成的CuZn合金是一类具有高活性的甲醇重整制氢催化剂,但同时ZnO在反应过程中的残余及其对CuZn合金性能的影响,包括催化剂失活是否是由于ZnO完全转化成Zn所导致,仍需要从原子尺度到微纳米尺度上的高分辨率进行表征和研究。
3.2 Cu/ZnO/Al2O3界面效应
前人研究发现通过增强活性位点的分散,可以实现催化剂性能的提高。Shokrani等[27]运用尿素燃烧法合成不同氧化铝负载比重的CuO/ZnO/Al2O3催化剂,结果表明,在传统CuO/ZnO体系内,氧化铝的加入会降低CuO和ZnO的相对结晶度,增大催化剂的比表面积[图11(a)],同时改善金属粒子的分散情况。掺入Al的比例过多会减少活性组分的Cu的占比,故氧化铝负载量存在最优值,当负载配比为C4Z4A2.5时催化剂催化性能最好[图11(b)]。

图11 不同Cu/Zn/Al质量比的CuO/ZnO/Al2O3纳米催化剂的比表面积(a)和甲醇转化率(b)[27]Fig.11 Specific surface area(a)and methanol conversion(b)of CuO/ZnO/Al2O3nanocatalyst with various Cu/Zn/Al mass ratios[27]
3.3 CuO/CeOx界面效应
表面氧物种和表面氧空位的共存对氧化还原过程有协同作用。一方面,甲醇的离解和氧化需要表面氧物种;另一方面,H2O的分解和H2的形成需要表面氧空位[68-70]。
以水煤气反应(WGS)为例,氧空位可以促进水和氧物种的反应,生成羟基,羟基再与一氧化碳进一步反应生成甲酸盐,然后甲酸盐分解生成二氧化碳和氢气。因此氧空位的生成可以降低重整产品中的一氧化碳含量,有效提高氢气产量[式(18)~式(22)[71-72]]。
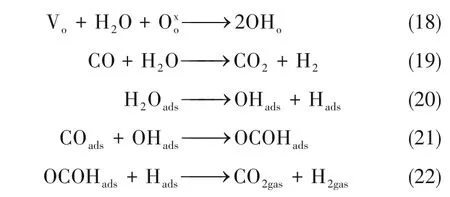
基于以上研究,有学者将容易形成氧空位的二氧化铈作为基底,通过构建富含氧空位的CuO/CeOx界面调控MSR的催化性能[69,71-74]。
Yang等[69]制备了纳米棒状(nano-rod,下文简称R)、纳米颗粒(nano-partical,下文简称P)和海绵状(sponginess,下文简称S)CeO2来负载Cu。由于CuO和CeOx之间的强相互作用,存在氧化还原反应[式(23)],Yang等认为Cu+的生成有利于提高催化活性,通过Cu+的峰面积与整个Cu LMM峰面积比值计算出表面Cu+相对含量[图12(a)、(b)],结果表明Cu+相对 含 量 顺 序 为CuO/CeO2-R>CuO/CeO2-P>CuO/CeO2-S,这与催化剂催化活性顺序基本一致。Yang等认为Ce4+到Ce3+的还原伴随着氧空位的产生,这有助于吸附氧转移到活性氧物种,晶格氧从本体迁移到表面,参与氧化还原反应。因此Ce3+含量越高,催化剂催化活性应越高,通过XPS表征发现CuO/CeO2-R具有最多的Ce3+[图12(c)],同时证实了与金属阳离子结合的晶格氧物种和吸附氧的存在[图12(d)]。具有最多Cu+和Ce3+的CuO/CeO2-R催化剂在260℃达到了100%的甲醇转化率,同时CO选择性为2.4%。

图12 CuO/CeO2-R,CuO/CeO2-P,CuO/CeO2-S等样品的Cu2p的XPS光谱(a),Cu LMM的Auger电子能谱(b),Ce3d的XPS光谱(c),O1s的XPS光谱(d)[69]Fig.12 XPS spectra of Cu2p binding energy region(a),Auger spectra of Cu LMM(b),XPS spectra of Ce3d binding energy region(c),XPS spectra of O1s binding energy region(d)of CuO/CeO2-R,CuO/CeO2-P,and CuO/CeO2-S[69]1—CuO/CeO2-R;2—CuO/CeO2-P;3—CuO/CeO2-S

Baneshi等[73]通过均相沉淀法制备了CuO-ZrO2-Al2O3、CuO-CeO2-Al2O3、CuO-ZrO2-CeO2-Al2O3催 化剂。通过X射线衍射(XRD)表征发现,向催化剂中添加CeO2降低了CuO特征峰的强度,表明CeO2改善了铜的表面分散并使铜粒径变小。通过FTIR红外光谱表征发现,CeO2掺杂的催化剂在3450cm-1处的加宽峰高于其他催化剂,这表明催化剂表面有较多OH物种,而OH物种的大量存在利于移除表面积炭,以提升催化剂稳定性。CuO-CeO2-Al2O3的甲醇转化率在240℃时达到100%,在此温度下未检测到CO,同时,CuO-CeO2-Al2O3催化剂显示出高寿命,其甲醇转化率在110h内保持100%不变。
Qiao等[74]在γ-Al2O3上原位生长合成M-Al-LDH(M=Mg、Ni、Co、Zn),而后煅烧成相应载体,并采用顺序浸渍法制备了一系列Cu-Ce/M-Al(M=Mg、Ni、Co、Zn)催化剂。与其他催化剂相比,Cu-Ce/Zn-Al催化剂具有最大的Cu比表面积和分散性,且还原温度最低,表面吸附氧的含量最高,具有最好的MSR性能。当反应温度为250℃时,Ce-Cu/Zn-Al的甲醇转化率达到100%,H2产率为779.7 cm3·kg-1·s-1。Chen等[45]制备了MOF衍生的CuO/CeO2催化剂,在543K下,该催化剂的H2产率为3648ml·(kg cat)-1·s-1,CO选择性为0.5%。
3.4 CuZnGaOx界面效应
Ga2O3同样可以以较高的分散度负载活性物质Cu。Ribeirinha等[75]开发了一种高活性和高选择性的CuO/ZnO/Ga2O3催化剂,该催化剂的低温MSR活性比来自Süd Chemie的市售CuO/ZnO/Al2O3催化剂的活性高两倍。催化剂的物理性质和反应的操作条件是影响其催化性能的重要因素,Ribeirinha等的研究表明100~250μm范围内的催化剂颗粒尺寸可使甲醇转化率最大化,将操作压力增加到0.1 MPa以上对甲醇转化是不利的,水醇比大于2时没有观察到显著的活性增加。
Ruano等[76]利用共沉淀法制备了CuZnGa(碳酸氢铵作沉淀剂)、CuZnGa-OH(氢氧化钠作沉淀剂)催化剂,阐明了MSR反应中铜的氧化状态,并将两催化剂性能和市售CuO/ZnO/Al2O3催化剂进行对比。对CuZnGa催化剂在反应初期下的质谱(MS)和近常压光电子能谱(NAP-XPS)的研究表明,铜纳米颗粒在反应的初始状态(TOS=4min)被氧化为Cu+,然后氧化物外壳快速还原为Cu0,同时将溶解氧物种保留在纳米颗粒的内层中[图13(a)],该亚表面氧物种会诱导表面铜物种变化生成Cuδ+,而Cuδ+生成有助于MSR催化活性的提高。Ruano等认为在CuZnGa样品上观察到的这种现象可能是由于铜的高分散性和催化剂存在表面缺陷位,而在化学成分相同但粒径较大、缺陷较少的CuZnGa-OH催化剂上未观察到这一现象。在180°C、催化剂用量与甲醇产量比(mcat/FMeOH)为150kg·s·mol-1的 反 应 条 件 下,CuZnGa有80.2%的甲醇转化率和99.5%的CO2的选择性,该值明显高于CuZnGa-OH(57.9%甲醇转化率)和Süd Chemie的 市 售CuO/ZnO/Al2O3催 化 剂(55.4%甲醇转化率)。
Tong等[77]通 过 共沉淀法将Ga掺 入Cu-Zn氧化物中,导致非化学计量的立方尖晶石相的产生,该立方尖晶石相包含间隙Cu+,可以在有缺陷的ZnGa2O4表面上原位产生大量极小的、高分散的0.5 nm铜簇[图13(b)、(c)],这可以抑制CO的生成,同时提高甲醇转化率。Tong等在pH=6.5 条件下制备的一系列不同配比的CuZnGaOx催化剂,在反应温度为150℃条件下检测MSR性能,发现所有催化剂CO检测含量都为0,但甲醇转化率较低。其中Cu/Zn/Ga配比为43/47/10的催化剂性能最好,甲醇转化率为22.5%。

图13 (a)CuZnGa样品H2释放的质谱检测以及对应时间下催化剂表面物种变化示意图[76];(b)CuZnGaOx还原后的HAADFSTEM图像:大铜颗粒(7~9nm)和小铜团簇(约0.5 nm,由白色箭头表示)的双峰分布[77];(c)还原后样品的原子探针层析成像(APT)数据显示固体基质中的高密度纯铜簇区域(红色斑块)[77]Fig.13 (a)The mass spectrometry detection of H2release from CuZnGa sample and the schematic diagram of the change of the catalyst surface species at the corresponding time[76];(b)HAADF-STEM image of CuZnGaOx catalyst after reduction showing bimodal distribution of large Cu particles(7—9nm)and small Cu clusters(~0.5 nm,indicated by white arrows)[77];(c)Atom probe tomography(APT)data of the specimen after reduction showing high density pure Cu cluster areas in the solid matrix(red patches)[77]
3.5 Cu/ZnO/ZrO2/Al2O3界面效应
Chang等[78]和Matsumura[79]的研究表明使用氧化锆作为促进剂可改善铜的分散性并防止铜铝尖晶石的形成。Agrell等[28]通过研究催化剂的抗反复氧化还原性能表明含有ZrO2的催化剂对氧化还原循环具有很高的耐受性,并在长期的实验中表现出较高稳定性,同时Agrell等提出在CuO/ZnO催化剂中添加Zr可促进Cu的还原。Patel等[80]采用湿浸渍法和共沉淀法制备了不同摩尔比的Cu/ZnO/ZrO2/Al2O3催化剂,探究ZrO2的加入对Cu/ZnO/Al2O3催化剂的影响,结果表明催化剂中掺入ZrO2增加了催化剂中Cu+的含量,提高了氢气的选择性,并降低了CO的产率,其中Cu/Zn/Zr/Al摩尔比为12/4/4/80的催化剂甲醇转化率可高达97%,CO在产物中占比0.8%(摩尔)。
Mateos-Pedrero等[81]利用水热法制备了不同Zr/Al摩尔比的CuZrAl催化剂,探究Zr/Al配比不同对催化剂物理性质及催化效能的影响。结果表明,当Zr/Al<1时可以获得成分均匀、Zr及Al物种高分散的催化剂,有利于提高MSR催化性能;当Zr/Al>1时,会形成具有不均匀表面和较低表面积的ZrAl载体,进而形成分散度较低、粒径较大的Cu颗粒,从而展现出较低的MSR活性;当Zr/Al比为0.4 时,催化剂具有最大的比表面积和Cu分散度,在220℃时进行MSR反应,产氢速率为460.1mmol·(g cat)-1·h-1,CO选择性为0.1%
Sanches等[82]采用共沉淀法制备了Cu/ZnO和Cu/ZnO/ZrO2催化剂,探究ZrO2的加入对Cu/ZnO催化剂结构和性能的影响。通过X射线衍射(XRD)、透射电镜(TEM)分析表明:Cu/ZnO/ZrO2催化剂中存在ZrO2纳米团簇,纳米团簇的存在可以阻止CuO和ZnO微晶生长,诱导Cu、Zn氧化物晶格发生微应变。这种效应可以促进更多暴露的CuO物种生成,使得铜更容易被还原,进而提升MSR性能。同时,在Cu/ZnO/ZrO2催化剂中还发现单斜ZrO2,单斜ZrO2对CO有较强的吸附能力,使得Cu/ZnO/ZrO2相较于Cu/ZnO催化剂有着更低的CO选择性。在250℃下测试两种催化剂的MSR性能,Cu/ZnO甲醇转化率为51.8%,CO选择性为4.7%,Cu/ZnO/ZrO2甲醇转化率为88.6%,反应中未检测到CO生成。
3.6 其他复合金属氧化物及其界面效应
Ploner等[83]用c-In2O3负载Cu,探究Cu-In2O3系统中的金属载体相互作用及二者的界面效应对MSR催化性能的影响。通过X射线衍射谱图(XRD)分析表明,将催化剂在0.1 MPa纯H2、300℃下预还原1h时,In2O3上形成金属铜颗粒[图14(a)],此时催化剂具有93%的CO2选择性,但甲醇转化率仅为56%;当其他条件不变,预还原温度提升到400℃后,可以检测到金属间化合物Cu2In的形成[图14(a)、(b)],此时催化剂CO2选择性达92%~94%,且表现出明显的自激活特性,从第一次到第四次连续运行中,甲醇转化率从62%提升到84%。Ploner等认为,金属氧化物系统中的相互作用可以用多步骤过程来描述,首先氧化物载体物种(In2O3)发生还原,随后还原的In2O3物种扩散到Cu晶格中,并形成金属间化合物[式(24)]。

图14 (a)Cu-In2O3体系在不同温度下进行氢还原后的不同形貌和性能示意图[83];(b)400℃氢还原1h后Cu/c-In2O3体系金属间化合物的透射电子显微镜分析[83]Fig.14 (a)Schematic diagram of different states and performances of Cu-In2O3system after hydrogen reduction at different temperatures[83];(b)Transmission electron microscopy analysis of the intermetallic compound state of the Cu/c-In2O3system after reduction in hydrogen at400°C for1h[83]

Lu等[84]首先通过共沉淀法制备CuZnAlOx(CZA)催化剂,再利用湿浸渍法将不同质量分数的硼(B)负载于CuZnAlOx上制得yB/CZA催化剂。通过BET、XRD、N2O化学吸附分析得:随着硼负载量的增加,催化剂的比表面积和Cu分散度先增加后降低,Cu的粒径随着硼负载量的增多先减小而后增大,存在最优硼负载量为0.3 8%,在250℃,水醇比为1/3,体积空速为9000ml·g-1·h-1时甲醇转化率为93%,一氧化碳选择性为0.3%。Kuo等[85]利用水热法制备了介孔的Cu-Fe/硅酸盐催化剂,其在180℃下甲醇转化率达99%。
Khani等[86]将Cu/γ-Al2O3、Cu/La-γ-Al2O3、Cu-Zn/La-γ-Al2O3、Cu-Zn/γ-Al2O3催化剂在不同温度下的产氢性能进行对比得出结论:在较高温度下,Cu/La-γ-Al2O3催化剂有相较其他催化剂更高的H2选择性和更低的CO选择性;而在较低温度下,Cu-Zn/γ-Al2O3表现出更为优异的催化性能;而当Zn、La同时存在时,则在所有温度范围都表现出最优秀的催化性能,认为Zn、La在不同的温度范围中可能对反应有不同的促进作用。Hwang等[87]通过自燃甘氨酸硝酸盐法制备了Cr、Fe不同摩尔比的CuCr1-xFexO2催化剂(x=0~1),通过BET、H2-TPR分析表明,随着Fe在催化剂中占比增加,催化剂表面积降低,还原峰温度降低,表明Cu与Fe之间的相互作用有利于催化剂还原,且有着较低还原温度的催化剂一般对应着较高的MSR活性[88]。
表3、表4统计了催化剂在不同制备方法、元素组成及比例条件下所导致的物理性质差异,以及在相应测试条件下的催化性能。可以看出通过尿素水解均相沉淀法和湿浸渍法制备得到的催化剂比表面积比其他方法的大。使用共沉淀法制备催化剂可以获得比较好的Cu的分散度。在催化剂中引入Ce、Zr、Ga可使得CO含量下降。温度在150~180℃下,催化剂的甲醇转化率普遍低于60%。CuO/CeO2在240~280℃下,甲醇转化率可以达到90%以上。CuO/CeO2-R在众多催化剂中表现出优异的性能,在260℃下,甲醇转化率100%,氢气时空收率15.2 1mol·(g cat)-1·s-1。

表3 Cu基催化剂物理性质汇总Table3 Summary of physical properties of Cu-based catalysts

续表3

续表3

表4 Cu基催化剂MSR催化效能汇总Table4 Summary of catalytic performance of Cu-based catalyst on MSR

续表4

续表4
4 Cu基催化剂失活原因
真实的工业生产过程中不仅对催化剂活性有要求,催化剂的稳定性优劣也是至关重要的,因此催化剂的失活是多相催化待解决的关键问题。而失活的原因主要有活性金属的团聚、焦炭的沉积、活性金属中毒等。
由Hughes[89]给出的金属稳定性的递增顺序Ag<Cu<Au<Pd<Fe<Ni<Co<Pt<Rh<Ru<Ir<Os<Re可见,Cu相较于其他金属稳定性较差,而且Cu的Hütig温度也较低,所以Cu相较于其他金属更易烧结。Kurtz等[90]对比了Cu/ZnO/Al2O3、Cu/ZnO和Cu/Al2O3的稳定性,发现加氧化铝作为结构助剂可明显抑制Cu晶体的热烧结。Zhang等[91]报道了CeO2可保持铜的高分散,Cu/CeO2/Al2O3在反应200h后甲醇转化率仍大于90.0%,而Cu/Al2O3催化剂在反应100h后迅速失活。Patel等[80]发现CeO2的掺入可产生流动氧来促进焦炭气化,以抑制焦炭沉积,Cu/ZnO/CeO2/Al2O3催化剂的积炭量相对于Cu/CeO2/Al2O3减少12。
Choi等[92]在铜/氧化锌/氧化铝催化剂运行的前100h运用X射线光电子能谱(XPS)观察到铜的还原,且当进料没有水时失活加剧[图15(a)],Choi等认为Cu2+到Cu0的转变是失活的主要原因。Thurgood等[93]从动力学角度研究,认为甲醇水蒸气重整反应中氢和含氧中间体分别占据一种活性位点,这同时也论证了在反应机理部分Frank等[39]的观点。通过测量两种位点浓度变化过程发现,氢吸附位点的浓度下降速度比吸附含氧中间体的位点要快,因此Thurgood等[93]认为氢吸附位点吸附、分解氢的能力下降是导致失活的原因。Liu等[94]将失活的Cu/CeO2催化剂用不同的还原条件进行比对,一种直接在氢气下进行还原,另一种先高温煅烧后再进行还原。结果表明直接在氢气条件下还原的催化剂活性并未恢复,而先高温煅烧后再进行还原的催化剂活性得到恢复[图15(b)、(c)]。因此,他们认为Cu的氧化不是催化剂失活的原因,而催化剂表面形成的碳质沉积[式(25)]是催化剂失活的主要原因。

根据Twigg等[95]的报道,硫对铜来说是一种强有力的毒物,式(26)的平衡常数约为1×105,因此来自H2S的硫或其他硫化合物会积聚在铜催化剂上,使得催化剂中毒[式(26)],而ZnO的存在可有效缓解硫中毒的现象[式(27)]。而氯化物可通过:①阻断或改变反应位点;②形成低熔点和高表面迁移率的氯化亚铜,加速烧结[式(28)];③加剧硫化物对Cu的中毒;④与氧化锌反应形成具有低熔点的卤化锌[式(29)],导致进一步的中毒和烧结问题。

5 结 论
本文对甲醇制氢Cu基催化剂及其反应机理、活性晶面、活性物种以及不同复合金属氧化物所产生的界面效应进行了综述和讨论。尽管MSR机理尚未产生一致性结论,但反应步骤大致有甲醇脱氢生成甲氧基,甲氧基脱氢生成CH2O*,水离解生成OH*或O*,氧物种与CH2O*进行偶联反应生成中间体,中间体进一步脱氢形成二氧化碳或一氧化碳;在偶联反应中,生成CO的路径具有最高的势垒,这可能是Cu基催化剂具有较低CO选择性的原因。对于甲醇水蒸气重整反应中的催化剂设计,首先由DFT计算得,Cu(110)晶面具有最高的活性和CO2选择性,所以在催化剂制备过程中应尽可能多地暴露该晶面。其次,氧空位作为吸附、转移氧物种的活性位点,可以有效促进含氧物种的反应,水可以在氧空位上解离生成羟基,羟基再与一氧化碳进一步生成甲酸盐,然后反应分解生成二氧化碳和氢气,可以有效降低重整产品中的一氧化碳含量,提高产氢效能。最后,优秀的Cu基催化剂应该有较大的Cu分散度和Cu比表面积,进而实现反应物与活性位点的充分接触,显著提升催化剂的质量活性。
针对Cu基催化剂中的氧空位和Cu基活性中心电子结构的调控,都可以利用Cu与不同金属氧化物间的界面效应来实现。例如Ce的掺杂可以构造氧空位,CuO与CeOx的相互作用还可以诱导产生Cu+;Ga在CuZn氧化物上的沉积可以产生高分散的铜簇,这可以抑制CO的生成;Zr在Cu/ZnO上的负载可以产生ZrO2纳米团簇,可以诱导CuZn晶格发生微应变促进Cu的还原;而In在Cu上的负载可以产生金属间化合物Cu2In,同时使催化剂具有自激活特性。同时,这些金属氧化物的引入,也可以起到分散Cu及活性位点的作用,因此通过继续深入研究和优化Cu基催化剂的界面效应对于提高甲醇水蒸气重整的反应效率至关重要。考虑到单个金属氧化物无法完全实现催化剂的活性、选择性和稳定性的全面调节,下一步的研究重点应该聚焦于发现或拓展一类能够协同各类金属氧化物与Cu的相互作用,使不同组分共同作为一个整体的催化剂,应用于甲醇水蒸气重整。
层状复合金属氢氧化物(LDHs)作为一类层板组成高度可控、层间具有可交换阴离子的二维纳米材料,其层板上高价态金属离子受静电斥力影响而呈现单分散的状态,这一独特性质为实现分散Ce、In等离子提供了保障。因此在以后的工作中,可以尝试以Cu、Zn作为LDHs层板的主要构建元素,选择性地向层板内掺杂In、Ce、Ga、Zr等金属阳离子,将这些具有良好界面效应的金属负载在活性金属Cu周围,然后煅烧形成三元或多元复合金属氧化物MMO,再运用H2还原,从而产生分散均匀的Cu0/Cu+离子和复合金属化合物,构建多种界面效应为一体的复合金属氧化物催化剂,从而制备出一类具有高活性、低CO选择性、高稳定性的甲醇水蒸气重整催化剂。在制备多元LDHs及其对应MMO的同时,目前鲜有研究关注层间具有不同氧化还原活性、离子半径的金属含氧酸根对于层板活性位点电子结构及焙烧产物MMO比表面积的影响,因此通过层板元素组成与分布、层间阴离子选择两个维度去实现对于LDHs及其焙烧产物MMO的电子结构、配位环境、活性位点分散度、催化剂表面酸碱性和比表面积的探索,将实现包括甲醇重整制氢反应在内的一系列多相催化反应催化剂的进一步优化。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
银行家(2022年5期)2022-05-24
福建农林大学学报(哲学社会科学版)(2021年5期)2021-12-04
法制博览(2021年14期)2021-11-25
市场周刊(2021年8期)2021-11-22
第一财经(2019年8期)2019-08-26
中南民族大学学报(自然科学版)(2019年1期)2019-04-04
安徽医科大学学报(2015年9期)2015-12-16
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
