
玉米盐胁迫相关性状全基因组关联分析及候选基因预测
2022-01-06 09:19单婷玉施雯王翌婷曹孜怡汪保华方辉
遗传 2021年12期
单婷玉,施雯,王翌婷,曹孜怡,汪保华,方辉
玉米盐胁迫相关性状全基因组关联分析及候选基因预测
单婷玉,施雯,王翌婷,曹孜怡,汪保华,方辉
南通大学生命科学学院,农业农村部南方平原玉米科学观测实验站,南通 226019
盐胁迫是影响玉米产量的重要因素,为探究玉米盐胁迫响应的遗传基础,本研究以150份遗传背景丰富的玉米自交系为材料,结合34,342个多态性SNP标记,利用混合线性模型对玉米两个盐胁迫相关性状进行全基因组关联分析。关联分析结果表明:共鉴定8个独立位点与盐胁迫相关性状显著关联,其中3个位点与枯萎度显著关联,分布在4号和9号染色体上,5个SNP位点与株高变化率显著关联,分布在1、2、3和6号染色体上。结合盐胁迫下基因的表达量数据和功能注释,筛选到11个候选基因,利用qRT-PCR验证其中7个基因在盐胁迫下表达量显著上调。本研究结果为玉米耐盐机理的解析奠定了基础,为玉米耐盐种质的培育提供新的靶基因。
玉米;盐胁迫;混合线性模型;全基因组关联分析
土壤盐碱是农业生产中面临的一个重要问题,盐胁迫对植物生长危害严重。植物体内高浓度的盐会造成渗透压失衡,植物根系很难吸收水分,并且高浓度的盐会造成植物离子中毒[1,2]。玉米(L.)具有高产、易管理、用途广泛等特性,深受人们喜爱,目前无论从种植面积还是产量,均高于水稻(L.)和小麦(L.),位于第一位。未来,玉米产量仍需进一步提升才能保障我国的粮食安全。然而,玉米是一种盐敏作物,很容易受到盐胁迫的影响,导致产量降低[3,4]。因此,在注重玉米产量持续提高的同时,也应关注抗逆玉米的培育。
早期研究表明,不同的玉米自交系在盐胁迫下展现出不同的盐胁迫反应[5],但对玉米盐胁迫响应的遗传基础研究仍然较少。QTL (quantitative trait loci)定位是一种经典的研究数量性状遗传结构的方法,植物中多种复杂性状都采用该方法进行遗传结构的解析,包括多种作物的耐盐性QTL定位[6~10]。例如,水稻中最早被克隆的QTL是[11],其编码HKT(high-affinity K+transporter)类型的蛋白,在盐胁迫下能维持Na+/K+的体内平衡。在玉米中,Cui等[12]用一个包含161个家系的重组自交系群体结合3000个单核苷酸多态性标记(single nucleotide polymorphism, SNP)验证了8个盐胁迫响应QTL。最近,Zhang等[13]利用Zheng58和Chang7-2组配的重组自交系群体成功克隆了基因,研究表明的功能缺失能增加叶片中Na+的积累,提高对盐离子的敏感性,进一步证明基因对盐胁迫的正向调控作用。
另外,全基因组关联分析(genome-wide association mapping, GWAS)也是解析复杂数量性状遗传结构的常用方法。Luo等[5]利用关联群体鉴定到57个位点与玉米盐胁迫响应显著关联,筛选了49个候选基因,其中44%的基因与胁迫响应相关,同时,利用转基因技术验证了和两个基因调控玉米的盐胁迫响应功能。除此之外,通过GWAS方法和基因也被相继克隆[14,15],分别编码HAK家族的离子转运蛋白和Ca+结合蛋白。综上所述,作为影响玉米产量的重要胁迫因素,无论在QTL还是基因水平,针对玉米耐盐性的研究还相对较少,仍然需要克隆更多的基因,从而更深入的了解玉米盐胁迫响应机制,辅助培育耐盐玉米品种。
本研究基于一个包含150个自交系的关联群体,利用混合线性模型,剖析玉米盐胁迫响应的遗传结构,挖掘玉米盐胁迫相关的候选基因,为玉米耐盐种质的选育奠定基础。
1 材料与方法
1.1 供试材料与表型鉴定
本实验所用关联群体共包含150份玉米自交系,主要是一些EX-PVP材料,或者是一些重要的公共自交系。这些自交系单行种植、自交授粉后,在成熟期收获,自然晾晒后保存种子用于表型鉴定。
每个自交系选15粒种子,置于100 mL三角瓶中,加入适量10%的H2O2淹没种子,不停地摇晃三角瓶,使种子充分消毒。10 min后将瓶中H2O2倒掉,用大量清水冲洗干净。种子消毒后在28℃的暗环境中培养两天以促进种子萌发,种子萌发后将其置于发芽纸上列成一排,卷起后竖着放置于装有适量水的塑料盒中,放置于人工气候室中继续培养。光照周期为白天12 h,温度28℃/20℃,相对湿度75%。待幼苗长至二叶期,去除胚乳,转移至霍格兰全营养液中继续培养至三叶期,随后设置对照组和处理组,对照组持续用霍格兰全营养液培养,处理组用200 mmol/L的氯化钠溶液处理,处理11 d后对两组幼苗的株高、枯萎度、鲜重和干重的表型进行鉴定,每个自交系测量4~6株。株高/干重/鲜重变化率=(对照–处理)/对照×100%。枯萎程度分为5个等级:I级,幼苗生长正常,无明显的盐害症状;II级,幼苗生长受到轻微抑制,种子尖端枯萎;III级,1~2片叶片呈黄色或褪绿;IV级,幼苗生长被显着抑制,只有心绿;V级,幼苗完全死亡[16]。
1.2 基因型鉴定
利用美国Illumina公司开发的MaizeSNP50 BeadChip芯片[17]对所有家系的基因型进行鉴定。该芯片包含56,110个SNP标记,涵盖19,350个玉米基因。利用plink1.5软件[18]分析每个标记的缺失率、杂合率和最小等位基因频率(minor allele frequency,MAF)。MAF≥0.05,缺失率和杂合率<20%的SNP标记被保留,最终有34,342个多态性位点用于后续分析。
1.3 群体结构、亲缘关系和邻接树构建
群体结构是在Linux系统下利用Admixture1.3软件[19]完成,设置亚群数量=2~10,计算交叉验证误差(cross-validation error, CV error),较好的值的选择能够产生较低的CV error。亲缘关系系数是使用Tassel5.0软件中Centered_IBS算法[20]计算得出。邻接树的构建,首先是利用Tassel5.0软件计算成对SNP标记之间的状态同源(identical by state, IBS),随后在Mega7.0软件中使用极大似然法计算[21]。
1.4 全基因组关联分析
利用Tassel5.0软件的混合线性模型(mixed linear model, MLM)[22],同时控制群体结构和亲缘关系,对两个耐盐性状进行全基因组关联分析。所用SNP标记的MAF≥0.05,阈值设置为<1.0×10–4 [5,15]。其统计模型为:
y =β+α+υ+µ+e
其中,y为表型观察值;β为标记和群体结构以外的未知固定效应值;α为标记的效应值;υ为群体结构的效应值;µ为多基因遗传背景的效应值;e为残差;为群体结构的矩阵;、、分别为y与β、υ和µ相关的矩阵。
1.5 上位性互作分析
每个位点最显著的SNP标记被用于上位性互作分析。双尾方差分析用于估计成对加性-加性的上位性互作[23,24],阈值设置为<0.05。上位性互作效应是通过比较包含所有单基因座效应的完整模型的残差与简化模型的双位点相互作用效应得出的。
1.6 候选基因预测及qRT-PCR验证
参考Li等[25]方法,在显著位点前后50 kb窗口内查找候选基因。随后在MaizeGDB网站(https:// www.maizegdb.org)搜索玉米盐胁迫诱导的基因表达数据库[26],获得这些基因盐胁迫下的基因表达情况,结合基因的功能注释,筛选候选基因。
LH196是一个来自美国的商业自交系,耐盐性较好。对其进行盐处理,方法与关联群体的处理方法一致,用200 mmol/L的氯化钠溶液处理长势一致的二叶期LH196幼苗,取处理后0 h和12 h的叶片,用RNA提取试剂盒(日本TaKaRa公司)提取RNA,用反转录试剂盒(日本TaKaRa公司)将RNA反转录成cDNA。使用ABI 7500实时定量PCR仪进行qRT-PCR检测,每个样品设置3个生物学重复和3次技术重复。利用2−ΔΔCt方法计算基因的表达量。玉米基因作为内参对照,文中所有基因引物序列详见附表1。
2 结果与分析
2.1 盐胁迫性状的表型数据分析
150份玉米自交系的盐胁迫表型数据来自文献[16]。4个盐胁迫相关表型中,枯萎度和株高变化率呈现出正态分布的趋势,变异广泛(附图1,附表2)。其中,枯萎度被划分为5个等级展现出丰富的变异,而鲜重和干重变化率呈现明显的偏分离(附图1,附表2),不适合利用混合线性模型进行分析。因此,后续分析中将鲜重和干重变化率去除。
2.2 群体结构和亲缘关系
为了探究150份玉米自交系之间的遗传关系,利用Admixture 软件的交叉验证程序(cross-validation procedure),结合34,342个SNP对这些自交系进行群体结构分析。每一个给定的值(假定的亚群的数量)计算出的交叉验证误差见图1A。=2时展现了最高的交叉验证误差,虽然=7时的交叉验证误差最低(0.88),但将该关联群体分成7个亚群并不符合当前育种群体的实际情况。当=3时,交叉验证误差发生急剧的下降,下降0.066,而=4~6时,下降程度缓慢,平均下降0.011,结合一个被广泛应用到复杂数量性状遗传基础解析中的关联群体对群体结构的划分[25,27],最终将这150份玉米自交系组成的关联群体划分为3个亚群(图1B),分别为硬杆群体(stiff stick, SS)、热带亚热带群体(tropical/subtropical, TST)和非硬杆群体(non-stiff stick, NSS)。=2时反应了初始的亚群分化,将以B73为代表的SS群体分化出,=3时,紧跟SS群体又分化出新的亚群,即TST群体。第3个亚群为NSS群体,包括SS群体之外的多数温带材料。综上所述,这个玉米关联群体被划分成3个亚群,其中SS群体包含46个自交系,NSS群体包含47个自交系,TST群体包含29个自交系。除此之外,还有28个自交系被划分成混合群体(Mixed亚群),因为这些自交系每个亚群的概率值都低于0.6 (附表3)。
邻接进化树的构建也展示了与群体结构相似的结果,只存在微小的差异(图2A)。进化树分成了3个分支,分别是SS、NSS和TST亚群,而28个来自Mixed亚群的自交系分布在整个进化树中。最小的分支包含36个自交系,包括全部29个TST亚群的自交系,1个NSS亚群的自交系和6个Mixed亚群的自交系。第二个分支包含45个自交系,包括39个NSS亚群的自交系和6个Mixed亚群的自交系。最大的分支由69个自交系组成,包括SS亚群的全部46个自交系,7个NSS亚群的自交系和16个Mixed亚群的自交系。
同样,主成分分析(principal component analysis, PCA)可以明显的将150份玉米自交系分成SS、NSS和TST三个亚群,而混合的亚群分布在三者中间(图2B),前两个主成分能够清晰的区分这几个亚群。其中,第一个主成分能够解释21.69%的遗传变异,反映了SS和NSS或TST群体的分化;第二个主成分可以解释3.89%的遗传变异,反应了NSS和TST的分化。

图1 150份玉米自交系的群体结构分析
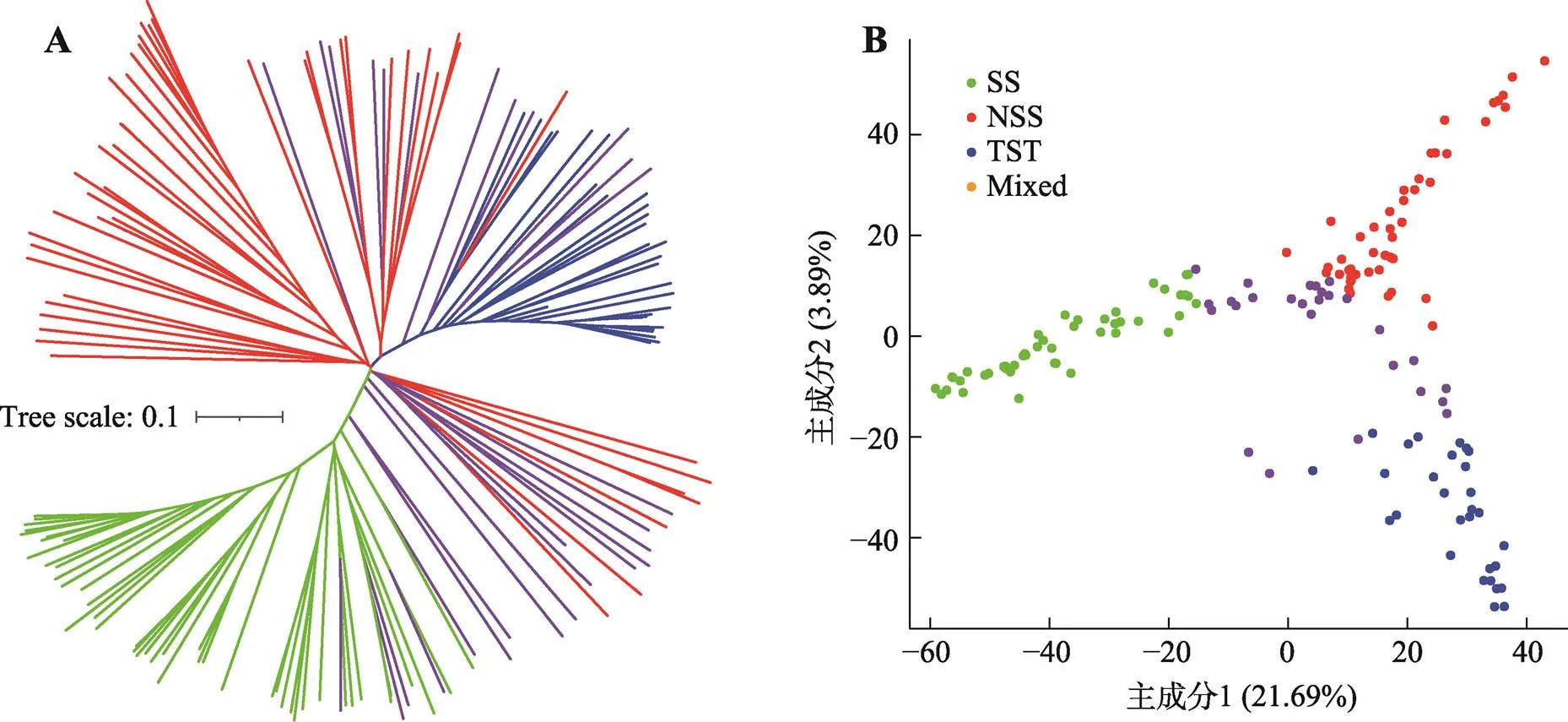
图2 150个玉米自交系的进化分析
除此之外,从亲缘关系的结果(表1)可以看出,28.13%的亲缘关系系数小于0.1,表明这些家系之间几乎没有亲缘关系。54.47%的亲缘关系系数分布于0~0.2之间,表明大部分家系之间的亲缘关系较远。30.63%的亲缘关系系数分布在0.2~0.4,展现了较弱的亲缘关系。另外,还有13.39%的亲缘关系系数分布在0.4~1.0之间,表明了小部分的自交系之间亲缘关系较近。剩余1.5%的亲缘关系系数大于1,表明极少数个体间相似性较高。总的来说,150份自交系之间,极少数个体之间展现了较高的相似性,大部分个体间的亲缘关系较远甚至没有。
2.3 混合线性模型分析
利用混合线性模型(MLM)对150份玉米自交系的两个盐胁迫性状(枯萎度和株高变化率)进行全基因组关联分析(图3:A,B)。枯萎度性状只检测到1个显著的位点,位于9号染色体上。株高变化率检测到8个显著的SNP,分别位于1、2、3和6号染色体上。根据这些显著SNP间的连锁不平衡程度将位于相同LD区域的SNP合并,发现两个性状分别鉴定到1个和5个显著的独立位点。将这些独立的显著位点作为协变量进行条件分析时,枯萎度性状检测到两个新的位点,均位于4号染色体上(图3:C,D)。综上所述,枯萎度性状共检测到3个显著的独立位点,株高变化率检测到5个显著的独立位点。利用一般线性模型估算这些位点解释的表型变异,发现与枯萎度关联的位点可以解释25.69%的表型变异,与株高变化率关联的5个位点可以解释34.92%的表型变异。除此之外,还对两个性状的显著位点进行上位性互作分析,发现在株高变化率这一性状中,鉴定到4对上位性互作,包括SYNGENTA5755与SYN1142、PHM13742.5和PZE-106065133之间互作,以及SYN1142与PHM13742.5之间互作,这些互作共可以解释5.24%的表型变异。因此,综合加性效应和上位性效应,株高变化率性状中,5个显著性位点共可解释40.16%的表型变异(表2)。
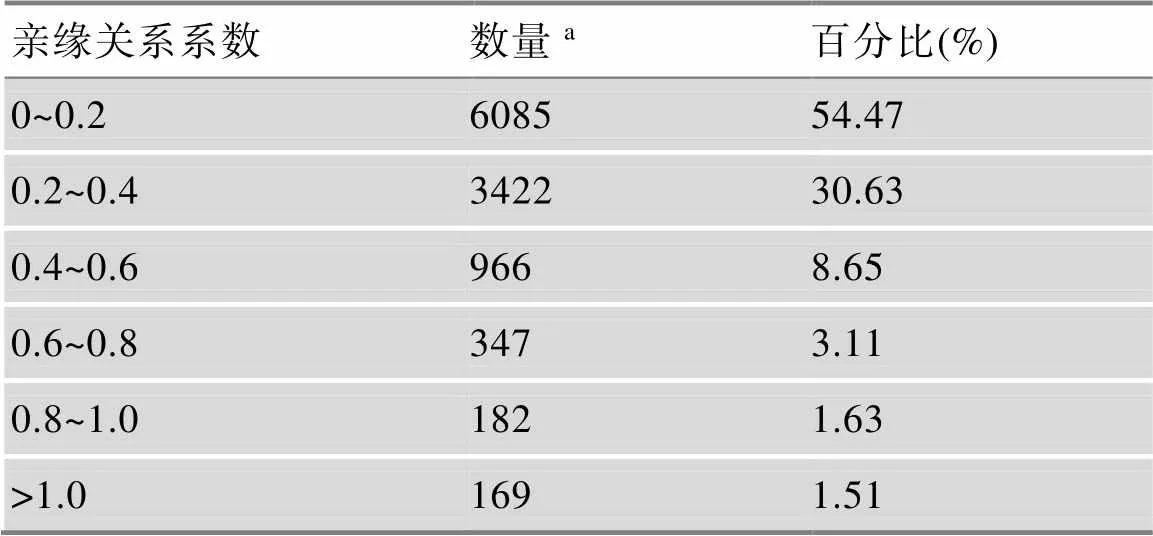
表1 亲缘关系系数统计分析
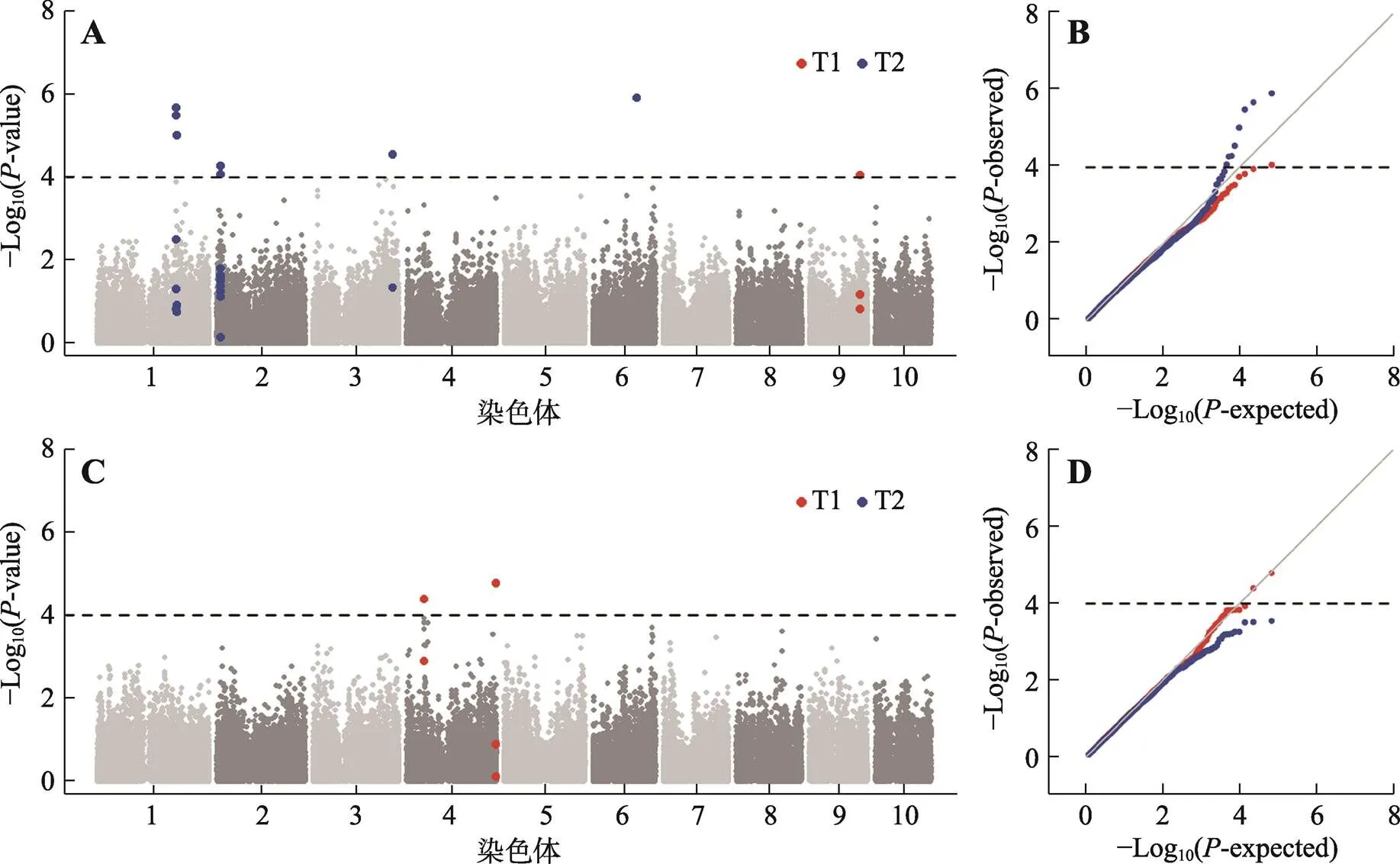
图3 基于混合线性模型两个盐胁迫性状的关联分析
2.4 候选基因分析
结合盐胁迫下基因的表达量数据和功能注释,在8个显著位点下鉴定到11个候选基因(表3,附表4),对这些基因进行GO富集分析(图4),发现4个基因编码酶或编码具有酶活性的蛋白:编码CASP-like蛋白,具有脱氢酶活性;编码包含泛素类结构域的蛋白(Ubiquitin-like domain-containing protein),具有ATP酶活性以及与分子伴侣结合的能力;编码5ʹ脱氧核苷酸酶(5ʹ-deoxynucleotidase);编码丝氨酸/苏氨酸蛋白激酶,催化磷酸从ATP转移到丝氨酸或苏氨酸残基。两个基因具有转录因子活性,其中,编码钙调素结合蛋白(Calmodulin-binding protein 60G),具有结合DNA的转录因子活性;编码MYB类型的转录因子(MYB family transcription factor),可以与特定的DNA序列结合,调控下游基因的表达。一个基因编码ABC转运蛋白(ATP-binding cassette transporter),可以转运无机离子、单糖、聚糖等多种物质。除此之外,还有4个基因编码蛋白,包括编码一个有缺陷的类泛素化蛋白(defective in cullin neddylation protein, DCN1-like protein 4),能够与泛素类蛋白结合酶结合;编码一个包含DUF1336结构域的蛋白,能够增强疾病抗性,具有防御或免疫功能,另外有两个基因和编码未知功能的蛋白。
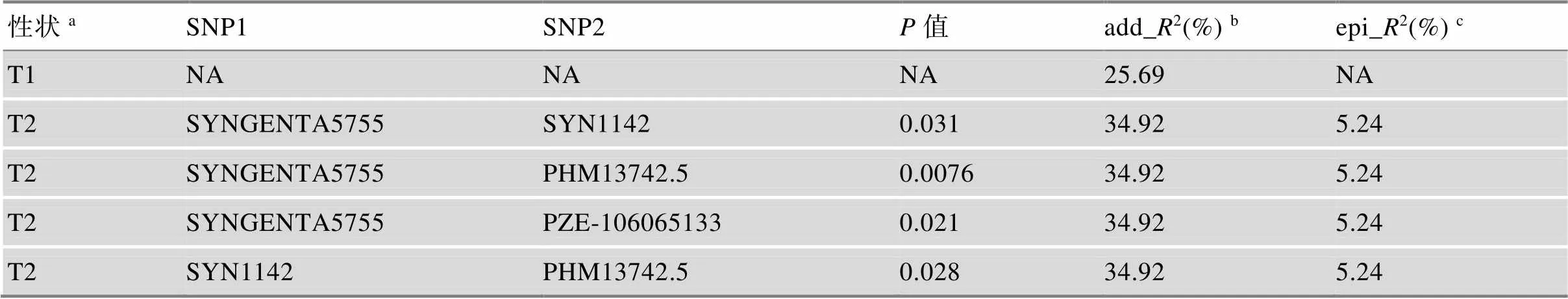
表2上位性互作分析结果
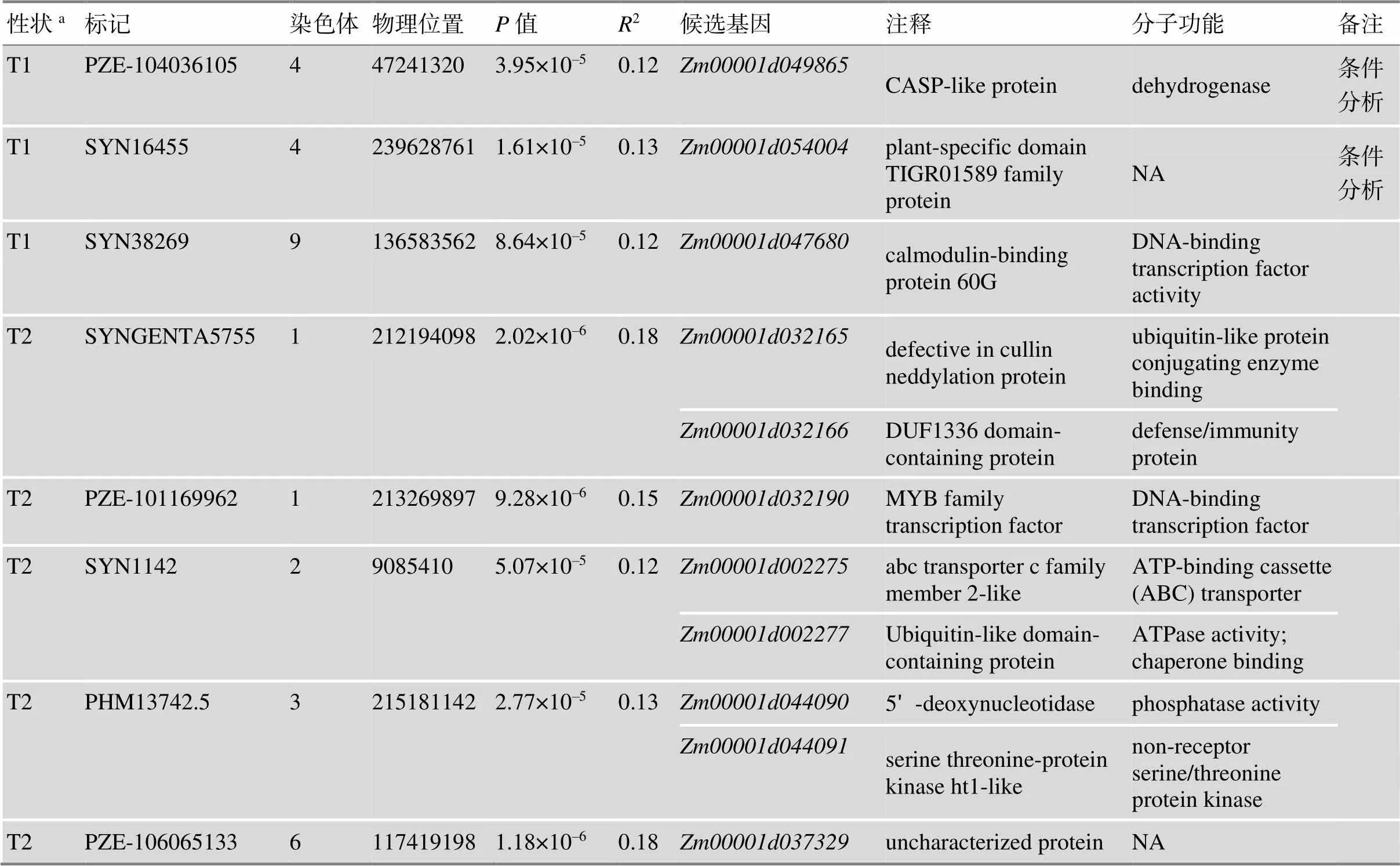
表3 两个玉米耐盐性状的全基因组关联分析结果

图4 盐胁迫候选基因的GO富集分析
2.5 盐胁迫下候选基因的表达验证
为进一步验证这些候选基因是否对盐胁迫有响应,利用qRT-PCR验证候选基因在盐胁迫0 h和12 h的玉米叶片中的表达情况,结果显示7个候选基因盐胁迫下表现出表达差异,其中3个基因的表达量呈现显著的差异(图5,<0.05),4个基因的表达量呈现极显著的差异(<0.01),而且这7个基因在盐胁迫下表达量均显著上调。其中,表达差异最大的是基因,盐胁迫12 h后其表达量升高了将近2倍(=0.0079)。该基因编码MYB类型的转录因子,可能在玉米抵抗盐胁迫中扮演重要角色。
3 讨论
本研究基于一个包含150份自交系的玉米关联群体,结合34,342个SNP标记,利用混合线性模型的方法,对两个盐胁迫相关表型进行全基因组关联分析,鉴定到8个显著的独立位点,同时筛选到11个候选基因。此外,利用qRT-PCR技术验证了7个候选基因在盐胁迫下差异表达,推测这些基因可能在玉米抵御盐胁迫中发挥作用。

图5 盐胁迫下候选基因的表达验证
3.1 一般线性模型与混合线性模型的结果比较
关联分析是剖析复杂数量性状遗传基础的重要手段,能够充分利用历史重组事件,极大提高定位精度[28]。一般线性模型和混合线性模型是关联分析常用的两个统计模型,因为关联分析容易受到群体结构和亲缘关系的影响[22,27],而混合线性模型同时控制群体结构和亲缘关系,能够更好的控制假阳性的现象,因此,应用更加广泛。
Xie等[16]基于同一个关联群体,利用一般线性模型对4个耐盐性状进行全基因组关联分析,共鉴定到7个位点与盐胁迫性状显著关联,这些位点分布在1、3及6号染色体上,其中4个位点与本研究的结果共定位。而基于混合线性模型的关联分析鉴定到4个新的位点,这些位点分布在2、4和9号染色体上(表4)。基于这4个新的位点筛选到5个候选基因,其中3个基因在受到盐胁迫后均显著上调。这些基因的鉴定,为玉米响应盐胁迫机理的解析和耐盐种质的培育奠定基础。
3.2 玉米耐盐候选基因分析
基因在表达水平上调控很多重要的生物学过程,转录因子在这一过程中发挥了重要作用[29,30],因此它们有望成为改良作物复杂数量性状的优良候选基因。MYB转录因子家族庞大,功能多样,普遍存在于真核生物中[31]。许多MYB基因已被证明参与植物的胁迫响应,例如盐胁迫和干旱胁迫。在拟南芥中异位过表达小麦的基因可以显著增强对干旱和盐的胁迫[32],进一步研究表明,该基因能够促进渗透压平衡重建和活性氧(ROS)清除能力。过表达小麦的另一个MYB基因——也能显著提高拟南芥的耐盐性[33]。同样,玉米和基因对盐胁迫有响应[34,35]。除了作物外,在多个木本植物中也有关于MYB转录因子耐盐胁迫的报道[36,37]。因此,MYB转录因子在盐胁迫响应中具有重要作用。本研究鉴定到一个MYB转录因子,qRT-PCR结果表明该基因在盐胁迫时具有最大程度的表达上调响应,该基因还未被克隆,很可能在玉米盐胁迫响应中行使功能,是耐盐种质改良的重要候选基因。除此之外,还有6个候选基因在响应盐胁迫时不同程度地上调表达,可能以直接或间接的方式参与玉米的盐胁迫响应。
3.3 与其他盐胁迫关联分析的比较
玉米中一个应用及其广泛的关联群体,包含508份自交系,已在玉米农艺和产量性状[38]、籽粒油份含量[25]、抗病[39]、玉米苗期抗旱[40,41]和盐胁迫相关性状[5,14,15,42]中被成功应用,显示了关联分析在解析复杂数量性状、挖掘相关基因上的优势。在利用该关联群体进行盐胁迫相关性状的研究中,采用了3种不同的表型收集方式,分别为盐处理后茎叶中NA+/K+的含量[14,15]、存活率[5]和株高、根长、干重、鲜重等性状[42],鉴定了多个显著关联的位点,并克隆了几个调控盐胁迫响应的基因,如、、和等。这些研究都用了超过100万个多态性位点,覆盖了基因组中绝大多数的基因,对盐胁迫响应的相关基因进行挖掘,分别鉴定到67~149个独立的位点,其中,解释的表型变异(explained phenotypic variation, PVE)超过15%的主效位点数极少,平均PVE低于10%,暗示了盐胁迫性状复杂的遗传基础。此外,有关盐胁迫相关研究中所鉴定到的候选基因均不相同,表明不同性状的遗传结构可能差异较大。本研究所用的关联群体包含150份自交系,所用标记数为34,342个,采用的统计方法与文献[5,14,15,42]中相同(都为MLM)。尽管在群体大小和标记数量上有差异,但本研究也定位到8个独立的位点,平均可以解释14.13%的表型变异,这可能与群体的背景差异有关。同时,筛选并初步验证了7个新的基因在盐胁迫时会上调表达,这一发现加强了对盐胁迫相关性状遗传基础的认识,并为耐盐种质的选育提供新的靶基因。

表4 两种关联分析统计模型结果的比较
附录:
附加材料详见文章电子版www.chinagene.cn。

附表1 qRT-PCR引物序列
附表2 150份自交系的分群情况总结

附表3 盐胁迫相关性状的表型结果

续附表3

续附表3
续附表3

附表4 显著SNP位点前后50k区间内的所有基因信息
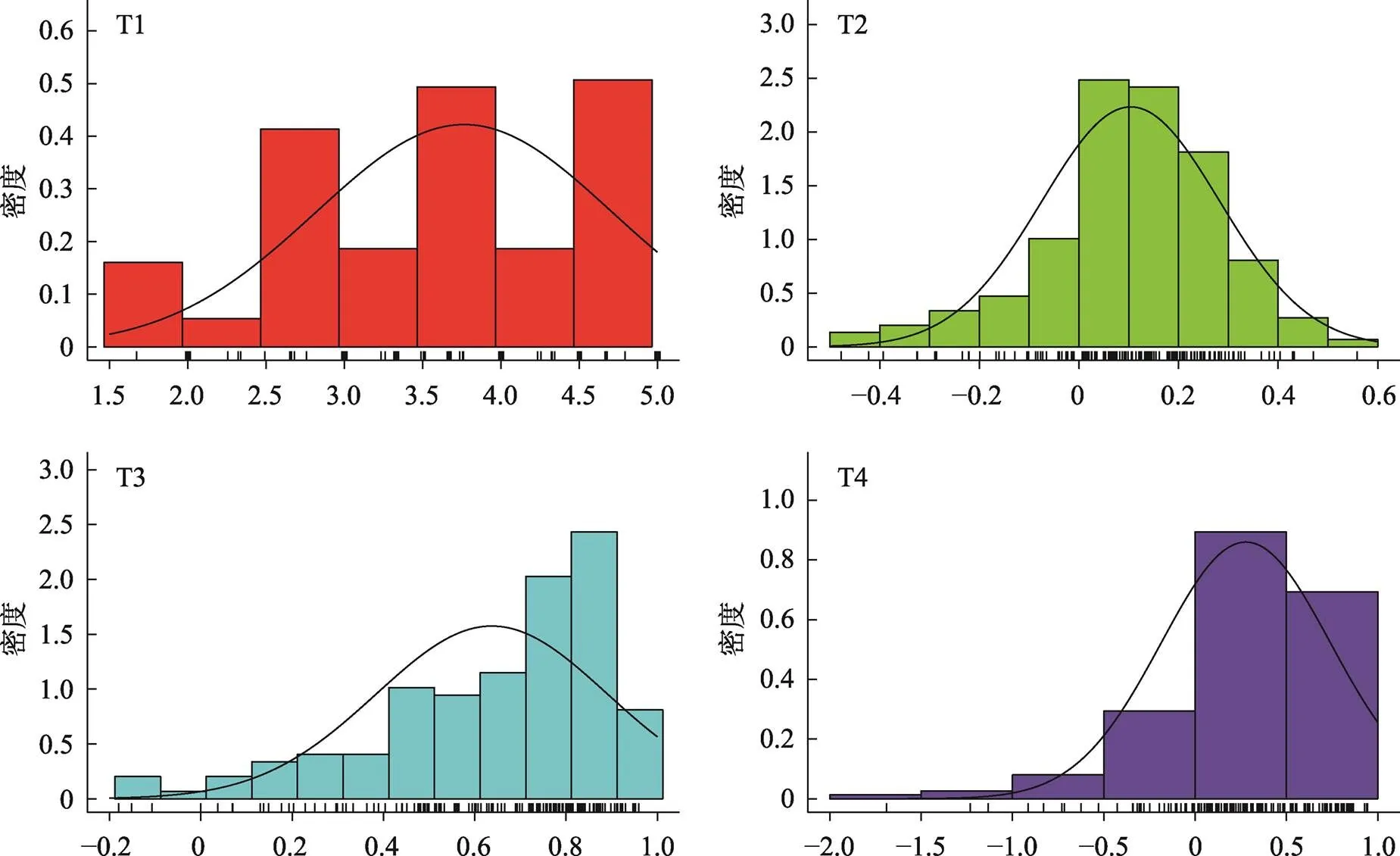
附图1 关联群体盐胁迫相关性状的表型分布
[1] Deinlein U, Stephan AB, Horie T, Luo W, Xu GH, Schroeder JI. Plant salt-tolerance mechanisms., 2014, 19(6): 371–379.
[2] Hu T, Zhang GX, Zheng FC, Cao Y. Research progress in plant salt stress response., 2018, 16(9): 3006–3015.胡涛, 张鸽香, 郑福超, 曹钰. 植物盐胁迫响应的研究进展. 分子植物育种, 2018, 16(9): 3006–3015.
[3] Munns R, Tester M. Mechanisms of salinity tolerance., 2008, 59: 651–681.
[4] Liang WJ, Ma XL, Wan P, Liu LY. Plant salt-tolerance mechanism: a review., 2018, 495(1): 286–291.
[5] Luo X, Wang BC, Gao S, Zhang F, Terzaghi W, Dai MQ. Genome-wide association study dissects the genetic bases of salt tolerance in maize seedlings., 2019, 61(6): 658–674.
[6] Prasad SR, Bagali PG, Hittalmani S, Shashidhar HE. Molecular mapping of quantitative trait loci associated with seedling tolerance to salt stress in rice (L.)., 2000, 78(2): 162–164.
[7] Hamwieh A, Tuyen DD, Cong H, Benitez ER, Takahashi R, Xu DH. Identification and validation of a major QTL for salt tolerance in soybean., 2011, 179(3): 451– 459.
[8] Ren ZH, Zheng ZM, Chinnusamy V, Zhu JH, Cui XP, Iida K, Zhu JK. RAS1, a quantitative trait locus for salt tolerance and ABA sensitivity in., 2010, 107(12): 5669–5674.
[9] Luo MJ, Zhao YX, Zhang RY, Xing JF, Duan MX, Li JN, Wang NS, Wang WG, Zhang SS, Chen ZH, Zhang HS, Shi Z, Song W, Zhao JR. Mapping of a major QTL for salt tolerance of mature field-grown maize plants based on SNP markers., 2017, 17(1): 140.
[10] Luo MJ, Zhang YX, Chen K, Kong MS, Song W, Lu BS, Shi YX, Zhao YX, Zhao JR. Mapping of quantitative trait loci for seedling salt tolerance in maize., 2019, 39(5): 1–12.
[11] Ren ZH, Gao JP, Li LG, Cai XL. Huang W, Chao DY, Zhu MZ, Wang ZY, Luan S, Liu HX. A rice quantitative trait locus for salt tolerance encodes a sodium transporter., 2005, 37(10): 1141–1146.
[12] Cui D, Wu D, Somarathna Y, Xu CY, Li S, Li P, Zhang H, Chen HB, Li Z. QTL mapping for salt tolerance based on SNP markers at the seedling stage in maize (L.)., 2015, 203(2): 273–283.
[13] Zhang M, Cao YB, Wang ZP, Wang ZQ, Shi JP, Liang XY, Song WB, Chen QJ, Lai JS, Jiang CF. A retrotransposon in an HKT1 family sodium transporter causes variation of leaf Na+exclusion and salt tolerance in maize., 2018, 217(3): 1161–1176.
[14] Zhang M, Liang XY, Wang LM, Cao YB, Song WB, Shi JP, Lai JS, Jiang CF. A HAK family Na+transporter confers natural variation of salt tolerance in maize., 2019, 5(12): 1297–1308.
[15] Cao YB, Zhang M, Liang XY, Li FR, Shi YL, Yang XH, Jiang CF. Natural variation of an EF-hand Ca2+-binding- protein coding gene confers saline-alkaline tolerance in maize., 2020, 11(1): 186.
[16] Xie YH, Feng Y, Chen Q, Zhao FK, Zhou SJ, Ding Y, Song XL, Li P, Wang BH. Genome-wide association analysis of salt tolerance QTLs with SNP markers in maize (L.)., 2019, 41(10): 1135–1145.
[17] Ganal MW, Durstewitz G, Polley A, Bérard A, Buckler ES, Charcosset A, Clarke JD, Graner E, Hansen M, Joets J, LePaslier M, McMullen MD, Montalent P, Rose M, Schön C, Sun Q, Walter H, Martin OC, Falque M. A large maize (L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome., 2011, 6(12): e28334.
[18] Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses., 2007, 81(3): 559–575.
[19] Alexander D, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation., 2011, 12(1): 1–6.
[20] Bradbury PJ, Zhang ZW, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples., 2007, 23(19): 2633–2635.
[21] Kumar S, Tamura K, Nei M. MEGA: molecular evolutionary genetics analysis software for microcomputers., 1994, 10(2): 189–191.
[22] Yu JM, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckle ES. A unified mixed- model method for association mapping that accounts for multiple levels of relatedness., 2006, 38(2): 203–208.
[23] Yu SB, Li JX, Xu CG, Tan YF, Gao YJ, Li XH, Zhang Q, Maroof MAF. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid., 1997, 94(17): 9226–9231.
[24] Wen WW, Liu HJ, Zhou Y, Jin M, Yang N, Li D, Luo J, Xiao YJ, Pan QC, Tohge T, Fernie AR, Yan JB. Combining quantitative genetics approaches with regulatory network analysis to dissect the complex metabolism of the maize kernel., 2016, 170(1): 136–146.
[25] Li H, Peng ZY, Yang XH, Wang WD, Fu JJ, Wang JH, Han YJ, Chai YC, Guo TT, Yang N, Liu J, Warburton ML, Cheng YB, Hao XM, Zhang P, Zhao JY, Liu YJ, Wang GY, Li JS, Yan JB. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels., 2013, 45(1): 43–50.
[26] Hoopes GM, Hamilton JP, Wood JC, Esteban E, Pasha A, Vaillancourt B, Provart NJ, Buell CR. An updated gene atlas for maize reveals organ-specific and stress-induced genes.2019, 97(6): 1154–1167.
[27] Yang XH, Gao SB, Xu ST, Zhang ZX, Prasanna BM, Li L, Li JS, Yan JB. Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize., 2011, 28(4): 511–526.
[28] Yang XH, Yan JB, Zheng YP, Yu JM, Li JS. Reviews of association analysis for quantitative traits in plants., 2007, 33(4): 523–530.杨小红, 严建兵, 郑艳萍, 余建明, 李建生. 植物数量性状关联分析研究进展. 作物学报, 2007, 33(4): 523– 530.
[29] Meng YY, Wang ZY, Wang YQ, Wang CN, Zhu BT, Liu H, Ji WK, Wen JQ, Chu CC, Tadege M, Niu LF, Lin H. The MYB activator WHITE PETAL1 associates with MtTT8 and MtWD40-1 to regulate carotenoid-derived flower pigmentation in medicago truncatula., 2019, 31(11): 2751–2767.
[30] Wang J, Zhou L, Shi H, Chern M, Yu H, Yi H, He M, Yin JJ, Zhu XB, Li Y, Li WT, Liu JL, Wang JC, Chen XQ, Qing H, Wang YP, Liu GF, Wang WM, Li P, Wu XJ, Zhu LH, Zhou JM, Ronald PC, Li SG, Li JY, Chen WX. A single transcription factor promotes both yield and immunity in rice., 2018, 361(6406): 1026–1028.
[31] Ambawat S, Sharma P, Yadav NR, Yadav RC. MYB transcription factor genes as regulators for plant responses: an overview., 2013, 19(3): 307–321.
[32] Qin YX, Wang MC, Tian YC, He WX, Han L, Xia GM. Over-expression of TaMYB33 encoding a novel wheat MYB transcription factor increases salt and drought tolerance in., 2012, 39(6): 7183–7192.
[33] He YN, Li W, Lv J, Jia YB, Wang MC, Xia GM. Ectopic expression of a wheat MYB transcription factor gene, TaMYB73, improves salinity stress tolerance in., 2012, 63(3): 1511–1522.
[34] Chen YH, Cao YY, Wang LJ, Li LM, Yang J, Zou MX. Identification of MYB transcription factor genes and their expression during abiotic stresses in maize., 2018, 62(2): 222–230.
[35] Wu JD, Jiang YL, Liang YN, Chen L, Chen WJ, Cheng BJ. Expression of the maize MYB transcription factor ZmMYB3R enhances drought and salt stress tolerance in transgenic plants., 2019, 137: 179– 188.
[36] Dong W, Liu XJ, Li DL, Gao TX, Song YG. Transcriptional profiling reveals that a MYB transcription factor MsMYB4 contributes to the salinity stress response of alfalfa., 2018, 13(9): e0204033.
[37] Guo HY, Wang YC, Wang LQ, Hu P, Wang YM, Jia YY, Zhang CR, Zhang Y, Zhang YM, Wang C, Yang CP. Expression of the MYB transcription factor gene Bpl MYB46 affects abiotic stress tolerance and secondary cell wall deposition in betula platyphylla., 2017, 15(1): 107–121.
[38] Yang N, Lu YL, Yang XH, Huang J, Zhou Y, Ali F, Wen WW, Liu J, Li JS, Yan JB. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel., 2014, 10(9): e1004573.
[39] Li N, Lin B, Wang H, Li XM, Yang FF, Ding XH, Yan JB, Chu ZH. Natural variation inconfers banded leaf and sheath blight resistance in maize., 2019, 51(10): 1540–1548.
[40] Wang XL, Wang HW, Liu SX, Ferjani A, Li JS, Yan JB, Yang XH, Qin F. Genetic variation incontributes to drought tolerance in maize seedlings., 2016, 48(10): 1233–1241.
[41] Mao HD, Wang HW, Liu SX, Li ZG, Yang XH, Yan JB, Li JS, Tran LP, Qin F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings., 2015, 6: 8326.
[42] Luo MJ, Zhang YX, Li JN, Zhang PP, Chen K, Song W, Wang XQ, Yang JX, Lu XD, Lu YX, Zhao JR. Molecular dissection of maize seedling salt tolerance using a genome-wide association analysis method., 2021, 19(10): 1937–1951.
Genome-wide association study and candidate gene prediction of salt tolerance related traits in maize
Tingyu Shan, Wen Shi, Yiting Wang, Ziyi Cao, Baohua Wang, Hui Fang
Salt stress is an important factor that affects maize yield. In order to explore the genetic basis of salt tolerance in maize, a genome-wide association study (GWAS) using the mixed liner model was conducted on 150 maize inbred lines with rich genetic background and 34,342 polymorphism SNP markers. A total of 8 independent loci were identified that significantly associated with salt-tolerance traits, among which 3 loci were significantly associated with withering degree on chromosomes 4 and 9; and the remaining 5 loci were significantly associated with plant height change rate on chromosomes 1, 2, 3 and 6. Eleven candidate genes were identified according to the gene expression data under salt stress; and functional annotations verified 7 of them to be significantly up-regulated under salt stress by qRT-PCR. These findings lay a foundation for understanding the mechanism(s) of maize salt tolerance and provide new target genes for the breeding of maize salt tolerance germplasm.
maize; salt tolerance; mixed liner model; genome-wide association study
2021-08-13;
2021-10-02
南通市科技项目(编号:MS22020033)和南通大学人才引进项目(编号:135420609055)资助[Supported by the Science and Technology Project of Nantong City, China (No. MS22020033)] and Talent Introduction Project of Nantong University (No. 135420609055)]
单婷玉,在读硕士研究生,专业方向:玉米盐胁迫响应研究。E-mail: shan_0822@126.com
方辉,博士,讲师,研究方向:植物复杂数量性状的遗传解析。E-mail: fanghui8912@126.com
10.16288/j.yczz.21-298
2021/12/08 5:07:32
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20211207.1156.002.html
(责任编委: 宋任涛)
猜你喜欢
现代临床医学(2023年1期)2023-03-24
中国医药科学(2022年5期)2022-05-05
北京农学院学报(2019年1期)2019-02-22
现代园艺(2017年21期)2018-01-03
河南农业科学(2017年4期)2017-04-12
西南农业学报(2016年5期)2016-05-17
西南农业学报(2016年6期)2016-04-16
中国卫生标准管理(2015年3期)2016-01-14
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
