
呋喹替尼在晚期结直肠癌中的研究进展
2022-01-06 07:10:00孙利英毛野黄志群黄胜兰李丹吴建兵
肿瘤防治研究 2021年12期
孙利英,毛野,黄志群,黄胜兰,李丹,吴建兵
0 引言
结直肠癌(colorectal cancer,CRC)是全球第三大常见的恶性肿瘤,也是导致死亡的最常见恶性肿瘤之一。2018年,全球约180万新发CRC病例,其中约88.1万例死亡[1-2]。从2013年到2018年,我国年龄标准化的CRC发病率从12.8/10万上升至23.7/10万,死亡率由5.9/10万上升至10.9/10万[3]。约25%的CRC患者在最初诊断时已经发生了转移,近50%的患者将发展为转移[4],肝脏是最常见的转移点。转移性结直肠癌(metastatic colorectal cancer,mCRC)的5年生存期为5%~15%[5]。晚期患者全身使用抗肿瘤药物,一线、二线标准治疗包括氟尿嘧啶、奥沙利铂、伊立替康、贝伐珠单抗和西妥昔单抗。然而,很多接受二线标准治疗后的患者,进展仍然是常见的[6],迫切需要有效的三线及以上的线路治疗。呋喹替尼由和记黄埔医药(中国医药科技(Chi-Med))开发,由于其在晚期结直肠癌的临床试验中显示出了良好的生存获益,该药于 2018年9月4日被国家食品药品监督管理局(China Food and Drug Administration,CFDA)批准上市,用于治疗至少经过二线系统化疗的复发或难治性晚期结直肠癌。本文综述了呋喹替尼的分子结构、作用机制、药代动力学、临床疗效和安全性,并讨论了其在其他肿瘤类型中的潜在临床应用。
1 分子结构与作用机制
呋喹替尼的化学名称是6-(6,7-二甲氧基喹唑啉-4-基氧基)-N,2-二甲基苯并呋喃-3-甲酰胺,分子式为C21H19N3O5,化学结构式见图1。呋喹替尼是一种具有强效型的新一代小分子酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI),抑制VEGFR1、2和3的IC50值分别为33、35和 0.5 nmol/L,对RET、FGFR-1和c-kit激酶有弱抑制作用(IC50:128~458 nmol/L),对其他激酶活性均无明显抑制作用(IC50>1 000 nmol/L),证实呋喹替尼对VEGFR1、2和3具有高度选择性[7-8]。这有望在维持靶点抑制的同时,最大限度地减少脱靶毒性,并在平均空闲时间(mean down time,MDT)提供高的药物暴露。
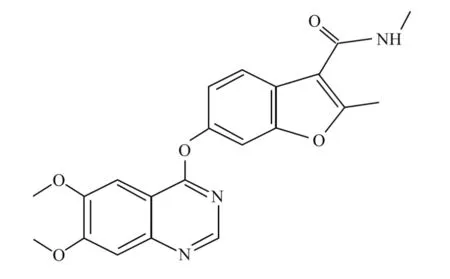
图1 呋喹替尼分子结构式Figure 1 Chemical structure of fruquintinib
呋喹替尼是一种通过抑制血管内皮细胞表面VEGFR磷酸化和下游信号转导,主要靶向VEGFR激酶家族(VEGFR1、2和3)的小分子。呋喹替尼通过抑制血管内皮细胞的增殖、迁移和管腔形成抑制肿瘤血管生成,从而发挥其抑制肿瘤生长的作用[7-8]。
呋喹替尼在分子水平可抑制VEGFR激酶的活性;在细胞水平,它抑制人脐静脉内皮细胞(human umbilical vein endothelial cells,HUVEC)和人淋巴内皮细胞(human lymphatic endothelial cells,HLEC)中VEGF/VEGFR家族激酶和VEGF/VEGFR细胞信号通路,IC50在低纳摩尔水平。在鸡胚绒毛尿囊膜(chick embryo chorioallantoic membrane,CAM)模型中阻断HUVEC功能,产生抗血管生成作用[7];在组织水平上,呋喹替尼可明显抑制CAM模型新生微血管的形成,口服呋喹替尼可显著抑制小鼠肺组织中VEGF诱导的VEGFR2磷酸化[9],VEGFR2磷酸化抑制的范围和持续时间与药物暴露密切相关[7];在整体动物水平上,呋喹替尼口服后可抑制VEGFR2/3磷酸化,抑制肿瘤血管形成,从而抑制肿瘤生成[10]。呋喹替尼采用一日一次给药,对结直肠癌以及其他多种类型肿瘤模型的生长均显示强效且剂量依赖性的抑制效应,在多种肿瘤敏感模型上可发现肿瘤缩小和消退[11]。在人肿瘤异种移植模型中,抗血管生成作用具有较强的抗肿瘤疗效,且具有良好的剂量反应。此外,在PDX模型中,呋喹替尼与具有良好耐受性的化疗药物联合使用时抗肿瘤活性增强,用呋喹替尼联合化疗药物多西紫杉醇或奥沙利铂时,可在患者来源的xenograph模型中发现肿瘤生长抑制增强[11]。异种移植模型中也观察到协同抗肿瘤效应,当呋喹替尼与其他靶向治疗(包括EGFR TKIs gefitinib、theliatinib和c-MET抑制剂savolitinib)联合使用时,可发现肿瘤生长抑制增强[12],作用机制见图2。

图2 呋喹替尼作用机制图Figure 2 Molecular targets of fruquintinib
2 药代动力学
2.1 吸收
健康受试者单次口服5 mg呋喹替尼胶囊,平均血浆药物峰浓度(Cmax)为155 ng/ml,达峰时间(Tmax)中位数为3 h(1.5~24 h),平均血浆药物浓度时间曲线下面积(area under the curve,AUC 0-∞)为5 700 h.ng/ml[13]。晚期癌症患者单次口服5 mg呋喹替尼胶囊,平均Cmax为195 ng/ml,Tmax中位数为2 h(0.5~2 h),平均AUC 0-72为 5 495 h.ng/ml[14]。
呋喹替尼在1~6mg剂量范围内暴露量(AUC)基本随剂量按比例增加,晚期癌症患者每日1次连续服药14天后,呋喹替尼暴露可达稳态,5 mg时平均稳态暴露率(AUC ss)约为首次给药暴露量(AUC 0-24)的3倍[14]。与空腹状态相比,健康受试者高脂餐单次口服4 mg呋喹替尼胶囊,Cmax下降约17%(几何均值比为82.9%,90%CI:0.767~0.89),AUC 0-∞相似(几何均值比为97.2%,90%CI:0.940~1.004)[13,15]。
2.2 分布
体外试验结果显示,呋喹替尼与人血浆蛋白结合率约为80%[7]。单次口服5 mg呋喹替尼,健康受试者和晚期癌症患者的口服消除相表观分布容积均值分别为32.5 L和42.2 L[13-14]。
2.3 代谢
C14标记的呋喹替尼体内代谢与物料平衡研究显示,呋喹替尼在人血浆中主要以原形存在,约占血浆中总暴露量的72%,经细胞色素P450酶3A(Cytochrome P450 enzyme 3A,CYP3A4)介导的去甲基代谢产物约占血浆中总暴露量的17%。其他代谢途径包括多位置单氧化、O-去甲基、N-去甲基、O-去喹啉唑环、酰胺键水解。Ⅱ相代谢产物主要是Ⅰ相产物的葡萄糖醛酸和硫酸结合物[13],代谢途径见图3。
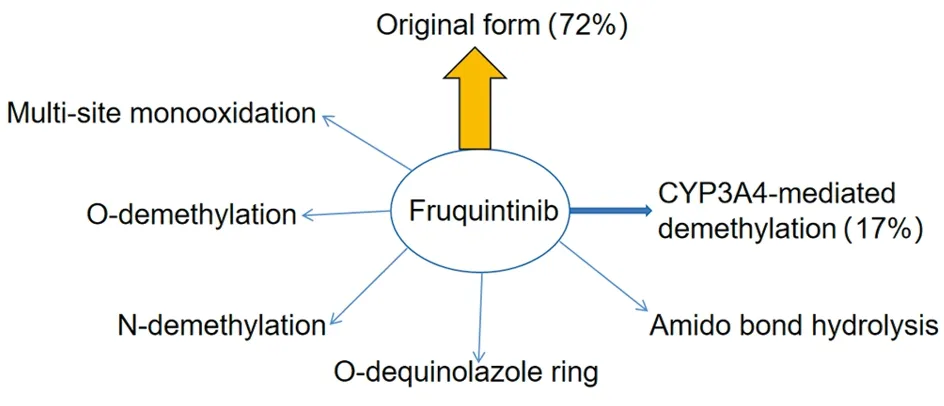
图3 呋喹替尼代谢途径图Figure 3 Metabolic pathway of fruquintinib
2.4 排泄
晚期癌症患者单次口服2~6 mg呋喹替尼,平均消除半衰期为35.2~48.5 h,平均口服清除率为9.98~17.8 ml/min[14]。健康受试者口服5 mg C14标记呋喹替尼,336 h内的放射性物质累积回收率平均为90.1%,其中尿液为60.3%(原形药为0.5%),粪便为29.8%(原形药为5.3%)。呋喹替尼主要经肾脏以代谢物形式随尿液排泄[13]。
2.5 药物相互作用
目前尚无药物相互作用的临床资料。呋喹替尼体内主要以原形存在,少部分经CYP3A4代谢,预计CYP3A4抑制剂或诱导剂对呋喹替尼的体内暴露影响有限。体外试验中未见呋喹替尼对CYP2C8、CYP2C9、CYP2C19、CYP2D6或CYP3A4的抑制作用,未见呋喹替尼对CYP1A2或CYP3A4的诱导作用[8]。
3 临床研究
早期的小分子VEGFR抑制剂,如瑞戈非尼[16]、贝伐珠单抗[17]、雷莫芦单抗[18]、舒尼替尼[19]和索拉非尼[20]具有较差的激酶选择性。他们为多靶点抑制剂,以相似的效力抑制超过10种激酶。因此,最大耐受剂量(maximum tolerated dose,MTD)的药物暴露由于多个靶点的抑制而受到限制,导致所有靶点,特别是VEGFR的抑制较不理想或抑制持续时间较短。而呋喹替尼是一种有效且高选择性的VEGFR1、2、3酪氨酸激酶抑制剂,有望最大限度地减少脱靶毒性,并在MTD时提供高剂量药物暴露,导致与生物制剂类似的持续VEGFR靶点抑制。由于靶点毒性分布狭窄,联合治疗具有灵活性[7]。
3.1 第Ⅰ阶段研究
在两家医院(NCT01975077)中进行了开放标签阶段单臂Ⅰb试验,42例mCRC患者给予口服呋喹替尼5 mg/d,连续21天,28天周期内休息7天。结果表明呋喹替尼具有良好的疗效。中位无进展生存期(median progression free survival,mPFS)为5.8月。中位总生存期(median overall survival,mOS)为8.88月。疾病控制率(disease control rate,DCR)为76.2%,客观缓解率(objective response rate,ORR)为9.5%[21]。
3.2 第Ⅱ阶段研究
在8家医院(NCT02196688)进行的一项随机、双盲、安慰剂对照的Ⅱ期临床试验中,71例患者按2:1的比例随机分配接受呋喹替尼或安慰剂[21]。主要终点是PFS,呋喹替尼组中位PFS为4.73月,安慰剂组中位PFS为0.99月(HR=0.30,95%CI:0.15~0.59,P<0.001),疾病控制率分别为68.10%和20.80%(P<0.001)。尽管呋喹替尼治疗50%以上的患者出现不同程度的肿瘤缩小,但mOS(7.72vs.5.52月,HR=0.71,P=0.29),ORR(2.10%vs.0.00%,P=0.45)未增加,可能是由于本试验样本量小,导致结果统计学差异不显著。
3.3 第Ⅲ阶段研究
FRESCO试验(NCT02314819)[22]是一项多中心(中国28家医院)、随机、双盲和安慰剂对照的Ⅲ期试验,比较了呋喹替尼与安慰剂治疗标准化疗失败的转移性结直肠癌患者。患者按2:1比例分为呋喹替尼组(n=278)和安慰剂组(n=138)。呋喹替尼组给予口服呋喹替尼5 mg/d,连续21天,28天周期内休息7天,并联合最佳支持治疗。对照组给予相同治疗周期的安慰剂口服联合最佳支持治疗。曾接受过VEGF或EGFR抑制剂治疗的患者被纳入试验,但接受过VEGF抑制剂的患者被排除。患者继续治疗,直到肿瘤进展或无法忍受的毒性发作或研究退出。研究主要终点是mOS,接受呋喹替尼的患者中位OS为9.3月,而接受安慰剂的患者mOS为6.57月(HR=0.65,95%CI:0.51~0.83,P<0.001);作为次要终点,mPFS为3.71和1.84月(HR=0.26,95%CI:0.21~0.34,P<0.001)。与安慰剂组相比,呋喹替尼组ORR也显著增加(4.7%vs.0,P=0.01)和DCR(62.2%vs.12.3%,P<0.001)。在亚组分析中,曾接受VEGF抑制剂(P=0.72)或者EGFR抑制剂(P=0.75)治疗与否无统计学意义,而K-RAS状态野生型与突变型差异有统计学意义(P=0.26)。
在FRESCO(一项随机、双盲、Ⅲ期临床试验)[23]中,对既往抗VEGF或抗表皮生长因子受体(epidermal growth factor receptor,EGFR)靶向治疗进行亚组分析,在既往使用靶向治疗(previous use of targeted therapy,PTT)和非PTT亚组患者中,呋喹替尼比安慰剂降低了37%的死亡风险。在非PPT亚组患者中,经呋喹替尼治疗的患者与安慰剂组患者的mOS为10.35vs.6.93月(P=0.003);在PTT患者中,呋喹替尼组与安慰剂组的mOS为7.69vs.5.98月(P=0.012)。在没有接受过抗VEGF治疗的患者中(可能包括接受过抗EGFR治疗的患者),呋喹替尼治疗的患者mOS更长(10.35vs.6.93,P<0.001)。对于曾接受过抗VEGF治疗的患者,呋喹替尼组与安慰剂组的mOS分别为7.20和5.91月(P=0.066),虽然差异未达到统计学意义,但可能是由于该亚组样本量较小。与安慰剂组相比,非PTT组和PTT组呋喹替尼组的PFS显著延长(P<0.001)。未接受抗血管内皮生长因子靶向治疗的患者(3.81vs.1.84月,P<0.001)和既往接受抗血管内皮生长因子靶向治疗的患者(3.48vs.1.84月,P<0.001)均能显著改善PFS。与安慰剂相比,服用呋喹替尼的患者在PTT组(3.6vs.0,P=0.303)和非PTT亚组(5.4vs.0,P=0.032)中显示出更高的ORR。与对照组相比,呋喹替尼组获得疾病控制的患者比例明显更高。
另外一篇回顾性研究[24]也提出既往使用抗VEGR药物治疗的患者比未使用抗VEGF药物治疗的患者的mPFS短(1.9vs.3.7月,P=0.006),而两组mOS无显著差异(9.0vs.8.5月,P=0.992)。此篇回顾性研究文中提出中性粒细胞淋巴细胞比值(neutrophil lymphocyte ratio,NLR)升高(HR=4.221,95%CI:1.683~10.586,P=0.002)是OS的独立预后因素。也有文章报道呋喹替尼和抗程序性死亡受体1(programmed cell death protein 1,PD-1)的组合可以协同抑制CRC进展,并改变肿瘤微环境,以支持抗肿瘤免疫反应[25]。
2019年CSCO会议口头汇报中提出,根据现有数据,与安慰剂相比,接受呋喹替尼治疗的肝转移患者OS、PFS显著改善;并且肝转移患者中,呋喹替尼组的肝毒性与安慰剂组差异无统计学意义。这对于临床用药具有指导意义。值得注意的是,呋喹替尼的其他临床试验也在进行中,总结见表1。
4 不良反应
在Ib期研究(NCT01975077)[21]中,42例受试者均发生了与治疗相关的不良事件(AEs),其中59.5%为3~4级AEs,3级及以上AEs以高血压(21.4%)、手足皮肤反应(hand and foot skin reaction,HFSR)(9.5%)、腹泻(9.5%)最为常见。有1例患者(2%)死于可能与治疗相关的致命性咯血。需要通过剂量调整(治疗中断或减少剂量)的常见AEs分别为血小板减少(11.9%)、HFSR(11.9%)和高渗(9.5%)。
在一项随机、双盲、安慰剂对照、多中心Ⅱ期临床试验(NCT02196688)[21]中,47例患者随机接受呋喹替尼。44例(93.6%)患者认为AEs与治疗相关。与治疗相关的3级及以上最常见(发生率5%)的AEs是高血压(29.8%)和HFSR(14.9%)。呋喹替尼组有3例参与者(1例伴有上消化道出血,1例胆红素升高,1例咯血)死于AEs。12例参与者(25.5%)有严重不良事件(SAEs)。15例(34.0%)因AEs而中断治疗。在呋喹替尼组中,有13例(27.7%)参与者因AEs而减少了研究剂量。需要改进治疗的常见AEs为HFSR(17.0%)、高血压(12.8%)和腹泻(4.3%)。
在中国Ⅲ期研究(FRESCO)[22]和对FRESCO研究安全性和不良事件的分析[26]中,呋喹替尼组266例(95.7%)和安慰剂组97例(70.8%)至少发生了一次治疗期间出现的不良事件(treatment emergent adverse event,TEAE);平均相对剂量强度分别为92%和98%。在呋喹替尼组中,最常见的治疗相关的不良反应是高血压(55.4%)、HFSR(49.3%)和蛋白尿(42.1%)。46%的接受呋喹替尼治疗的患者发生了与治疗相关的3~4级AEs,而安慰剂组的发生率为7.3%。最常见的3~4级与治疗相关的AEs是高血压(21.2%vs.2.2%)、HFSR(10.8%vs.0)和蛋白尿(3.2vs.0);上述事件发生的中位时间分别为10、21和20天。按年龄、性别和BMI进行的亚组分析显示,所有亚组中与治疗相关的TEAEs频率相似,并且与呋喹替尼的总体安全性一致。47.1%的呋喹替尼患者发生了需要治疗改变(治疗中断或剂量减少)的AEs,最常见的导致治疗改变的AEs是HFSR(13.3%)、蛋白尿(9.7%)和血小板计数下降(5.4%)。15.1%服用呋喹替尼的患者出现了导致停药的AEs。
FRESCO研究提出了生存质量调整的无疾病或毒性症状时间(quality-adjusted time without symptoms of disease or toxicity,Q-TWiST),一种客观整合生活质量和总生存时间来评估患者的获益。结果显示,呋喹替尼组的Q-TWiST明显长于安慰剂组(7.01月vs.4.78月),且无论之前是否接受过靶向治疗(抗VEGF或抗EGFR),患者均能从呋喹替尼中获益。结果表明,呋喹替尼不仅延长了患者的OS和PFS,并且在控制疾病同时保持良好的耐受性[27-30]。在2018年CSCO会议中Xu等提出,高血压事件与服用呋喹替尼疗效无明显关系。呋喹替尼在晚期NSCLC患者中的安全性和耐受性与结直肠癌患者一致[21-22]。
5 三线治疗药物
呋喹替尼、瑞戈非尼和TAS-102是晚期结直肠癌的三线治疗药物,但是临床上缺乏相关头对头的临床研究。有研究通过meta分析间接比较结果显示,呋喹替尼与瑞戈非尼、TAS-102疗效与安全性无明显差异[31-36]。在评估呋喹替尼与瑞戈非尼作为中国mCRC三线治疗的成本-效果时发现,呋喹替尼是一种具有成本效益的三线治疗方案,可节省约75 599元(11 454美元)[27]。
6 联合用药
2021年的ASCO会议上报道了两项关于呋喹替尼联合PD-1抑制剂在晚期结直肠癌中的研究进展,一项为呋喹替尼联合信迪利单抗在晚期结肠直肠癌和其他实体肿瘤的Ⅰb/Ⅰ期、多中心、两阶段研究[37]。截至2021年1月5日,共纳入44例经过一线、二线标准治疗耐药的CRC患者,中位随访时间8.3月,mPFS为6.8月,最常见的不良反应是蛋白尿(45.9%)和TSH升高(37.8%),导致呋喹替尼或信迪利单抗停用的事件各3例(5%)。呋喹替尼联合信迪利单抗在治疗晚期结直肠癌中显示出良好的疗效和安全性。另一项为呋喹替尼联合杰诺单抗(GB226)治疗转移性结直肠癌Ⅰb期研究[38],共纳入15例患者,其中1例为微卫星不稳定(microsatellite instability,MSI)型,12例为微卫星稳定(microsatellite stability,MSS)型,2例微卫星状态未知。在所有可评估的mCRC患者中,总ORR为26.7%,ORR为33.3%,DCR均为80%,mPFS为7.33月;在12例MSS型mCRC患者中,ORR为25.0%,DCR为75%,mPFS为5.45月。呋喹替尼联合杰诺单抗(GB226)治疗转移性结直肠癌显示出具有可控的安全性和较好的抗肿瘤活性。
7 其他瘤种的临床试验
呋喹替尼不仅用于治疗mCRC,也用于其他实体肿瘤,如肺癌、胃癌、胆道癌和乳腺癌。呋喹替尼已注册的临床试验见表1。

表1 呋喹替尼临床试验总结Table 1 Clinical trials of fruquintinib
对于晚期实体瘤,四项关于呋喹替尼的临床试验进行了注册,第一项为呋喹替尼在实体肿瘤、结直肠癌和乳腺癌中的多中心、开放性研究,用于评估呋喹替尼的药物代谢动力学(pharmacokinetics,pk)、安全性和耐受性的Ⅰ期试验(NCT03251378),这是呋喹替尼在美国患者中的首次临床试验。第二项是韩国进行的多中心、开放标签的Ⅱ期研究评价替雷利珠单抗(PD-1抑制剂)联合呋喹替尼治疗特定实体肿瘤的有效性和安全性(NCT04716634),这是呋喹替尼在亚洲非中国地区的首个临床试验。第三项是开放标签Ⅰb/Ⅱ期研究(NCT03903705),目的是评估呋喹替尼联合信迪利单抗注射液用于晚期实体瘤患者的安全性、耐受性、药代动力学和疗效。第四项是呋喹替尼在晚期实体肿瘤患者中的安全性和药代动力学Ⅰ期研究(NCT01645215),目前此试验患者招募工作已经完成,预计试验结果将于近期公布。
在肺癌方面,呋喹替尼有三项已经完成和一项正在进行的临床试验,其中一项已完成的是Ⅱ期临床试验(NCT02590965)[39],该试验是一项随机、双盲、安慰剂对照、多中心Ⅱ期临床试验,评价呋喹替尼加最佳支持治疗在晚期非鳞状非小细胞肺癌患者中的疗效和安全性。作为主要终点,与安慰剂相比,接受呋喹替尼的患者mPFS显著改善(3.8vs.1.1月,HR=0.34,P<0.001);作为次要终点,3月和6月生存率(90.2%和67.2%vs.73.3%和58.8%)、DRR(13.1%vs.0,P=0.041)和DCR(60.7%vs.13.3%,P<0.001)差异有统计学意义。呋喹替尼具有可接受的安全性和耐受性,最常见的3~4级TEAEs为高血压(8.2%)、HFSR(4.9%)和蛋白尿(4.9%)。第二项已完成的是FALUCA Ⅲ期试验(NCT02691299)[40],这项试验是为了进一步评估呋喹替尼在527例非鳞状非小细胞肺癌患者中的有效性和安全性。呋喹替尼组的mOS为8.9月,安慰剂组为10.4月(HR=1.02,95%CI:0.82~1.28,P=0.841),mPFS分别为3.7月和1.0月(HR=0.34,95%CI:0.28~0.43,P<0.001)。呋喹替尼的客观缓解率和疾病控制率分别为13.8%和66.7%,安慰剂为0.6%和24.9%(P<0.001)。在呋喹替尼治疗的患者中,高血压是最常见的治疗紧急不良事件(3级)(21.0%)。呋喹替尼与安慰剂相比延长了未接受后续抗肿瘤治疗的患者的中位总生存期(7.0vs.5.1月,HR=0.65,95%CI:0.46~0.91,P<0.012)。EORTC QLQ-C30和LC13问卷调查显示,接受呋喹替尼的患者大部分功能量表的生活质量也有改善。尽管这项研究没有达到其主要终点,但呋喹替尼联合其他药物治疗二线化疗失败的NSCLC患者可能有效。第三项临床试验旨在评估呋喹替尼联合吉非替尼用于EGFR突变的晚期非鳞非小细胞肺癌一线治疗的有效性和安全性(NCT02976116)[41],结果显示,作为主要终点,ORR为73.5%;作为次要研究,DCR为98.0%。两组的mPFS均为14.7月;外显子19缺失的患者PFS最高(16.5月,95%CI:12.9~21.2)。另一项还在进行中的临床研究为Ⅰb期研究(NCT03976856),旨在评估杰诺单抗(GB226)联合呋喹替尼治疗复发或转移性NSCLC患者的安全性和有效性。
在胃癌方面,已经注册了两项临床试验。一项是单臂Ⅰb/Ⅱ期试验(NCT02415023),目的是评估呋喹替尼联合紫杉醇治疗晚期胃癌的安全性、药代动力学特征和初步疗效。另一项试验(FRUTIGA;NCT03223376)是一项多中心、随机、双盲的Ⅲ期临床试验,旨在进一步评估呋喹替尼联合紫杉醇与紫杉醇单药二线治疗进展期胃癌的疗效和安全性。
在胆道癌方面,有评价呋喹替尼二线治疗晚期或转移性胆道癌的疗效和安全性的探索性研究(NCT04156958)。在乳腺癌方面,一项开放标签Ⅰb/Ⅱ期研究评估呋喹替尼联合替雷利珠单抗治疗晚期三阴性乳腺癌患者的安全性和有效性处于登记状态(NCT04577963)。
8 总结与展望
呋喹替尼是国产的VEGFR 1、2和3抑制剂,在治疗结直肠癌方面展现出了良好的疗效,已获CFDA批准上市。随着临床研究的不断扩展,对其他实体肿瘤进行的大多数试验仍在进行中,如肺癌、胃癌、胆管癌、乳腺癌,并且未来与化疗、免疫治疗及其他抗肿瘤疗法联合应用,可能会进一步提高疗效。目前,我们只有呋喹替尼在中国人群的临床试验数据,需要进行更多的临床试验来评估呋喹替尼在其他人群的疗效和安全性。到目前为止,仍然没有被证实的分子生物标志物来预测呋喹替尼的疗效。未来应该设计并实施旨在识别分子生物标志物以预测呋喹替尼疗效的试验。另一个关键问题是mCRC肿瘤对呋喹替尼耐药的未知机制,应该开展更多关于其耐药机制的研究,最重要的是如何逆转耐药。
猜你喜欢
Journal of Hainan Medical College(2023年15期)2023-12-06 07:54:42
基层中医药(2020年5期)2020-09-11 06:32:04
皮肤性病诊疗学杂志(2020年4期)2020-09-02 07:36:58
特别健康·下半月(2019年6期)2019-08-01 01:45:35
祝您健康(2019年3期)2019-03-22 08:57:08
时代英语·高一(2018年5期)2018-11-19 10:55:06
时代英语·高一(2017年5期)2017-11-14 15:52:20
中国卫生标准管理(2015年16期)2016-01-20 09:26:32
河南医学研究(2014年4期)2014-02-27 14:52:18
中国合理用药探索(2012年2期)2012-03-20 16:30:30
