
成骨分化内源性竞争性lncRNA-miRNA-mRNA网络与核心基因的研究
2022-01-06 13:25周子墨柳达陈森相覃森
中国骨质疏松杂志 2021年12期
周子墨 柳达 陈森相 覃森
中国医科大学附属盛京医院骨科,辽宁 沈阳 110004
成骨分化(osteogenesis differentiation)是一种涉及到多基因和多步骤的复杂过程。骨折、骨质疏松等骨病的发展和修复是成骨细胞和破骨细胞共同作用的结果。间充质干细胞(mesenchymal stem cell,MSCs)可以分化形成成骨细胞,成骨细胞通过基质分泌和钙矿化,促进骨质再生,起到重要的生物学作用[1]。
非编码RNA(noncoding RNA,ncRNA)是一类很少甚至没有编码潜能的RNA。在过去的研究中,其被认为是一类转录过程的“噪音”。随着高通量测序技术的发展,越来越多的noncoding RNA被识别靶向在mRNA表达的调控通路中,在调控核染色质结构和基因表达方面具有积极的意义[2]。长链非编码RNA(long noncoding RNA,lncRNA)是一类长度在200~100 000 nt的noncoding RNA。lncRNA被分为5种类型:正义型(sense)、反义型(antisense)、双向型(bidirectional)、内含型(intronic)、基因间型(intergenic),它们在功能上存在一定差异[3]。MicroRNA(miRNA)作为一类调控性RNA,可以通过多种方式介导基因表达,这些途径可以被上游的lncRNA靶向调控。目前,大量研究证明lncRNA可通过内源性竞争性RNA(ceRNA)学说靶向miRNA分子,形成miRNA海绵(Sponge)调节下游基因表达[4]。高通量测序技术可以通过基因芯片得到相应的差异基因,被广泛运用于基因组学和转录组学研究。Lim等[5]利用基因芯片,获得大量DEGs,预测了miRNA-mRNA潜在通路。
本研究通过对数据库中3个不同基因芯片对MSCs的成骨分化进行测序的结果进行分析,得到了差异表达的RNA及DEGs,通过靶点的互相预测,最终分析得到了差异较大的lncRNA-miRNA-mRNA互作网络,同时构建了PPI网络,预测关键基因,并将DEGs及关键基因进行功能富集分析和通路富集分析,为进一步的机制研究提供理论依据。
1 材料与方法
1.1 数据收集
从GEO公共数据库(https://www.ncbi.nlm.nih.gov/geo/)下载基因表达芯片GSE89330、GSE72429、GSE74837。GSE89330采用样本为成骨诱导分化14 d MSCs及分化的MSCs(对照组),平台为GPL16956,Agilent-045997 Arraystar human lncRNA microarray V3 (Probe Name Version);GSE72429采用样本为3个不同细胞系滑膜源性干细胞(synovial membrane-derived MSCs,SM-MSCs)和4个不同细胞系脂肪源性干细胞(adipose-derived stem cells,ADSC)来源的已分化成骨细胞,并与来自同一细胞系的未分化细胞进行比较。平台为GPL16770,Agilent-031181 Unrestricted_Human_miRNA_V16.0_Microarray (miRBase release 16.0 miRNA ID version)。GSE74837样本为用骨生成诱导方法诱导的2 d和5 d MSCs,对照组为未进行诱导的人骨髓间充质干细胞。平台为GPL13915 3 D-Gene Human Oligo chip 25k V2.1。
1.2 差异基因的数据分析
使用GEO2R(https://www.ncbi.nlm.nih.gov/geo/geo2r/)在线分析并获得差异基因(differentially expressed genes,DEGs)。GEO2R是美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)的GEO数据库自带公共在线分析工具[6],它将GEO数据进行limma差异表达分析,得出logFC、P.Value等数据。本研究将GSE74837得到的数据取P<0.05且∣logFC∣>2为DEGs;GSE72429得到的数据取P<0.05且∣logFC∣>1为DEmiRNAs;GSE89330得到的数据取P<0.05且∣logFC∣>2为DElncRNAs[7]。将得到的数据用火山图和热图表达,火山图使用R语言软件(版本1.3.1093)R包ggplot2绘制,热图使用TBtools软件(版本1.064)绘制。
1.3 基因功能富集分析
GO(gene ontology)是常用的DEGs功能作用分析方法,其可以通过注释基因或其产物、识别基因芯片数据而分析得到生物学特征结果。GO富集按照生物途径(biological process,BP)、细胞定位(cellular component,CC)、分子功能(molecular function,MF)对基因进行注释和分类。KEGG分析(Kyoto Encyclopedia of Genes and Genomes analysis)是另一种常用的基因通路分析方法,可以将基因注释和富集在信号通路上进行研究[8-9]。本研究应用DAVID (Database for Annotation,Visualization and Integrated Discovery)在线数据库分析工具进行分析,获得所需的GO和KEGG数据。本研究使用的DAVID数据库版本6.8,地址为https://david.ncifcrf.gov/,由美国国立变态反应与传染病研究所支持[10]。随后使用Fisher Exact 或EASE Score 统计方法,以GO数据各项P<0.05且FDR<0.05为筛选条件,KEGG<0.05为筛选条件进行分析筛选[11-12]。
1.4 内源性竞争性lncRNA-miRNA-mRNA互作用网络构建
内源性竞争性RNA(competing endogenous RNA,ceRNA)是以miRNA海绵为基础,lncRNA可直接与miRNAs相互作用并调控其活性的假设而构建的。基于这一假设,通过3个步骤建立了lncRNA-miRNA-mRNA网络:(1)筛选潜在可能的lncRNA、miRNA和mRNA。本研究以差异基因作为潜在可能靶点;(2)利用miRcode在线预测工具(http://www.mircode.org)根据mRNA预测潜在的miRNA靶点;(3)利用DIANA在线预测工具LncBase版本2.0(http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php)进行基于miRNA的潜在lncRNA靶点预测,筛选方法为临界值=0.7,细胞类型为hMSCs-BM,组织为骨组织,种类为干细胞[13]。最后,从三者中筛选出符合条件的RNA构建ceRNA网络。使用Cytoscape(version 3.5.1)可视化lncRNA-miRNA-mRNA网络。
1.5 蛋白互作网络构建和分析
STRING (Search Tool for the Retrieval of Interacting Genes)是一款在线分析工具,可以用来构建蛋白互作网络(protein-protein interaction network,PPI network)和进行在线分析[14]。在“Creative Commons BY 4.0”中,数据可以开放获取。将筛选的所有DEGs导入STRING(版本11.0,https://string-db.org/)在线分析软件,设置置信度(confidence score)≥0.4,互作最大值(maximum number of interactors)=0,得到相应数据[15],并导入Cytoscape(版本3.5.1)进行MCODE (Molecular Complex Detection,版本1.4.2)分析,设置参数为degree cutoff=2,node score cutoff=0.2,k-core=2,max.depth=100,从PPI中筛选出连接最为紧密的集簇,并将score设置为>4,得到两个最为紧密的集簇[16]。将得到的结果通过ClueGO+CluePedia进行GO富集分析。
1.6 关键基因筛选
将PPI网络导入Cytoscape(版本3.5.1)软件进行关键基因(hub gene)筛选,使用cytoHubba(版本0.1)进行分析及可视化,设置筛选条件hubba nodes ranked为degree=10,得到10个关键基因,并进行可视化处理[17]。
2 结果
2.1 差异表达RNA的筛选和鉴定
本研究选用基因表达芯片GSE89330、GSE72429、GSE74837,经GEO2R初步分析和条件筛选,共获得186个DEGs,包含81个下调基因和105个上调基因;89个DEmiRNA,包括25个下调miRNA和64个上调miRNA;441个DElncRNA,包括205个下调lncRNA和236个上调lncRNA。通过火山图和热图进行可视化,得到明显的差异表达(图1)。

图1 DEGs、DElncRNA、DEmiRNA的筛选和差异表达A:DEGs火山图,红色点代表筛选出的上调DEGs,蓝色点代表筛选出的下调DEGs;B:miRNA火山图,红色点代表筛选出的上调DEmiRNA,蓝色点代表筛选出的下调DEmiRNA;C:DElncRNA火山图,红色点代表筛选出的上调DElncRNA,蓝色点代表筛选出的下调DElncRNA;D:GSE74837中DEGs表达热图;E:GSE72429中DEmiRNA表达热图;F:GSE89330中DElncRNA表达热图。Fig.1 The screen and differently expression genes in DEGs, DElncRNA, and DEmiRNAA: In the DEGs volcano map, the red dot represents the screened up-regulated DEGs, and the blue dot represents the screened down-regulated DEGs; B: MiRNA volcano map, red dot represents the screened up-regulated demirna, and blue dot represents the screened down-regulated demirna; C: In the volcano diagram of delncrna, the red dot represents the screened up-regulated delncrna, and the blue dot represents the screened down-regulated delncrna; D: Heat map of DEGs expression in gse74837; E: Heat map of demirna expression in gse72429; F: Heat map of delncrna expression in gse89330.
2.2 GO功能与KEGG信号通路富集分析
根据GO分析和KEGG分析,以P<0.05,FDR<0.05为筛选条件,并按照计数值从大到小排列,将BP、CC与MF类别中排名在前10的富集结果用柱状图表示,将KEGG分析结果用气泡图表示(图2)。分析发现,在生物学途径上,DEGs主要富集在炎症反应和血管生成和骨骼系统发育中,促进MSCs成骨分化。在细胞成分中,DEGs大量富集在细胞膜和胞外体以及细胞外间隙。这提示DEGs表达分泌蛋白到胞外介导MSCs成骨分化。在分子作用上,生长因子活化、肝素结合等与细胞内外代谢、细胞生长和凋亡相关途径均被富集(图2A)。在KEGG分析中,发现DEGs主要富集在矿物质吸收、TNF信号通路、malaria等通路中(图2B)。
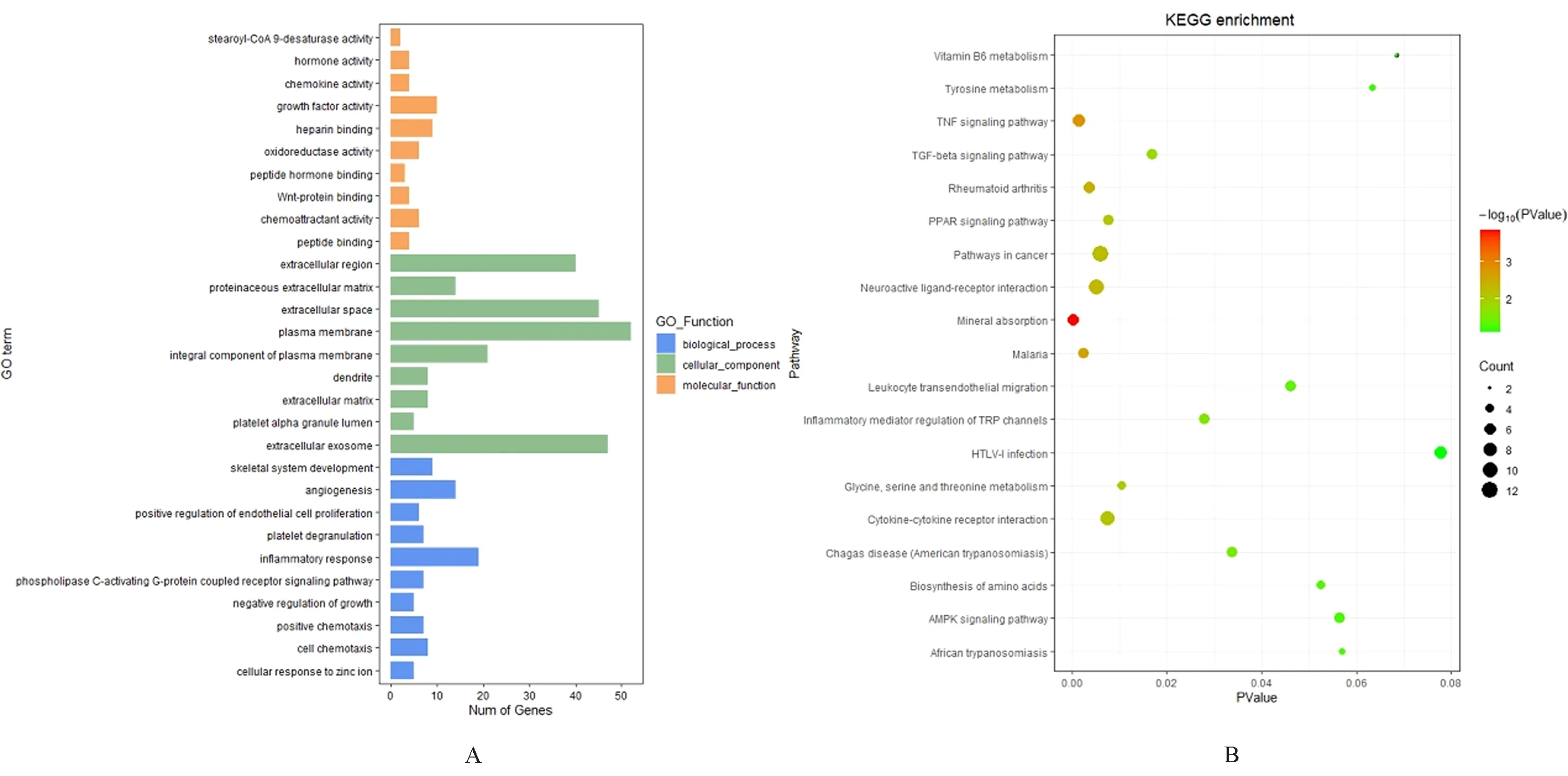
图2 DEGs GO富集分析与KEGG分析A:DEGs的GO富集分析;B:DEGs的KEGG分析。Fig.2 The GO enrichment analysis and KEGG analysis of DEGsA: Go enrichment analysis of DEGs; B: KEGG analysis of DEGs
2.3 内源性竞争性lncRNA-miRNA-mRNA互作用网络构建和分析
将筛选得到的DERNA进行比对和匹配,最终得到内源性竞争性lncRNA-miRNA-mRNA互作用网
络,并通过logFC值判断上下调,得到可视化网络(图3)。LncRNA SNAI3-AS1、TEX41、NR2F1-AS1和下游潜在靶点miRNA hsa-miR-1207-5p连接,DANCR和下游hsa-miR-1275连接,ARHGAP22-IT1和hsa-miR-224-5p连接,MEG8、TPT1-AS1、SNHG5和hsa-miR-335-3p,SNHG1和hsa-miR-1915-3p连接,LINC01503和hsa-miR-629-5p连接,LINC00472和hsa-miR-22-3p连接,调节下游DGEs表达。
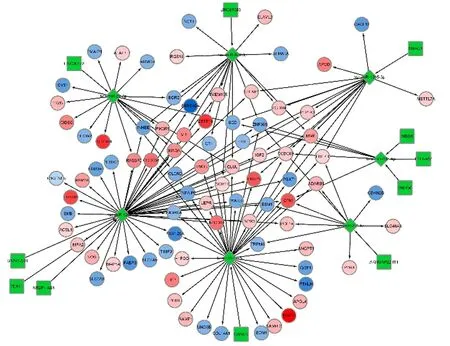
图3 内源性竞争性lncRNA-miRNA-mRNA互作用网络Fig.3 LncRNA-miRNA-mRNA network based on ceRNAs theory注:图中绿色方形代表lncRNA,绿色菱形代表miRNA,圆形代表DEGs,红色代表上调DEGs,绿色代表下调DEGs,颜色越深,∣logFC∣值越大。
2.4 蛋白互作网络构建及集簇模块筛选
通过STRING和Cytoscape软件分析与绘制,得出PPI网络(图4A)。红色点代表上调DEGs,绿色代表下调DEGs,颜色越深,∣logFC∣值越高。通过MCODE(Molecular Complex Detection,版本1.4.2),设置参数为degree cutoff=2,node score cutoff=0.2,k-core=2,max.depth=100,score>4,得到两个集簇模块,模块1包含14个节点(node),75条连线(edge),score=11.538(图4B);模块2包含6个节点(node),15条连线(edge),score=6(图4C)。富集分析结果显示,模块1中DEGs主要富集在白细胞血管内皮粘附(leukocyte adhesion to vascular endothelial cell)和中性粒细胞趋化(neutrophil chemotaxis)中(表1)。KEGG分析提示其主要富集于Malaria通路和类风湿性关节炎(rheumatoid arthritis)相关通路中。模块2中,DEGs主要富集在血管直径的正向调节(positive regulation of blood vessel diameter)中,KEGG分析中,主要富集在炎症介质对TRP通道的调节(inflammatory mediator regulation of TRP channels)通路中。

表1 蛋白-蛋白互作网络集簇DEGs的GO分析和KEGG分析Table 1 GO enrichment analysis and KEGG analysis in PPI network and significant clusters
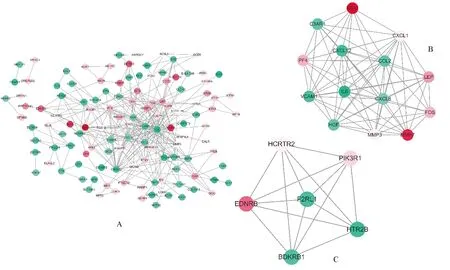
图4 PPI网络及集簇模块分析A:PPI网络构建,红色圆形代表上调的表达,绿色代表下调的表达,灰色线条代表相互连接;B:集簇模块(score=11.538),包含14个节点(node),75条连线(edge);C:集簇模块(score=6),包含6个节点(node),15条连线(edge)Fig.4 PPI network and significant clusters analysisA: PPI network construction, red circle represents up-regulated expression, green represents down-regulated expression, and gray line represents interconnection; B: Cluster module (score=11.538), including 14 nodes and 75 edges; C: Cluster module (score=6), including 6 nodes and 15 edges.
2.5 关键基因筛选
使用cytoHubba(版本0.1)进行筛选,获得了10个关键基因(hub gene)(图5A),颜色越深,代表ranked score排名越靠前。10个hub gene分别为IL6、CXCL12、CXCL8、CCL2、HGF、LEP、VCAM1、CXCL1、SAA1、FOS。对这10个hub gene进行GO分析,发现其主要富集在趋化因子活化、白细胞血管粘附和单核细胞趋化中(图5B、5D)。KEGG分析显示hub gene主要以Malaria相关通路和白血球粘附血管内皮相关通路为主(图5C)。

图5 基于degree方法在PPI网络中的前10个关键基因A:关键基因筛选及互作网络,颜色越深,代表ranked score排名越靠前;B:关键基因GO富集分析及法分类;C:KEGG富集分析及分类;D:关键基因GO富集直条图,纵坐标代表GO通路,横坐标代表富集的基因数目。Fig.5 Top 10 hub genes in PPI network ranked by Degree methodA: Key gene screening and interaction network, the darker the color, the higher the ranking of ranked score; B: Go enrichment analysis and classification of key genes; C: KEGG enrichment analysis and classification; D: Go enrichment bar graph of key genes, the ordinate represents go pathway, and the abscissa represents the number of enriched genes.
3 讨论
lncRNA和miRNA可以调节成骨分化中多种信号通路增强或抑制成骨分化能力[18-19]。本研究对GSE89330、GSE72429、GSE74837三组数据的分析和筛选,得到了lncRNA-miRNA-mRNA互作网络。Fang等[20]发现,DANCR可以通过调节滑膜液源性MSCs的miR-1275/MMP-13轴,诱导软骨形成。Chao等[21]通过对激素性股骨头坏死的患者miR-1207-5p的分析发现,患者miR-1207-5p水平显著升高,可能靶向抑制VEGF表达,抑制骨再生。本实验结果中同样获得了DANCR、miR-1207-5p、miR-1275等RNA的差异表达。这些潜在靶点可以为分子层面研究提供更多的理论依据。
通过对DGEs的GO分析及KEGG分析,发现DEGs富集在炎症反应、血管生成和骨骼系统发育中。在KEGG分析中,发现DEGs主要富集在矿物质吸收、TNF信号通路、malaria等通路中。在构建PPI互作网络后,通过集簇筛选和hub gene筛选得到最密集集簇和关键基因。通过再次GO分析和KEGG分析,将富集的范围缩小,得到最终的富集通路结果。关键基因主要富集在白细胞系带或滚动(GO:0050901)、白细胞血管内皮粘附(GO:0061756)及趋化因子活化(GO:0008009)等免疫相关通路中。成骨细胞可以产生免疫相关性蛋白和趋化因子,诱导B细胞的成熟。趋化因子CXCL12被证明在此过程中被上调,体内实验已经证明给予CXCL12后可以促进MSCs的聚集[22-24]。在本实验中,筛选出的关键基因中,CXCL12被证明显著下调,证明大量的趋化因子CXCL12靶向作用在MSCs细胞上。
综上,本研究通过构建lncRNA-miRNA-mRNA的互作网络,显示了SNHG1/miR-1915-3p/CXCL12、LINC00472/miR-22-3p/HSD11B1、LINC01503/miR-629-3p/ZBTB16、DANCR/miR-1275/MMP7等具有潜在靶向的通路。分析获得的相关差异基因IL6、CXCL12、CXCL8、CCL2、HGF、LEP、VCAM1、CXCL1、SAA1、FOS和ncRNA均在成骨分化中具有较高的组织特异性和功能特异性。
猜你喜欢
中老年保健(2022年1期)2022-08-17
清华金融评论(2022年4期)2022-04-13
口腔医学(2021年10期)2021-12-02
中学生数理化(高中版.高考理化)(2021年6期)2021-07-28
国际放射医学核医学杂志(2021年10期)2021-02-28
房地产导刊(2020年7期)2020-08-24
中华老年口腔医学杂志(2016年2期)2017-01-15
中国病理生理杂志(2015年8期)2015-12-21
天津护理(2015年4期)2015-11-10
中国药理学通报(2014年2期)2014-05-09
