
整合生物学先验信息的全基因组选择方法及其在家畜育种中的应用进展
2021-12-31 01:08袁泽湖李发弟乐祥鹏伟1
畜牧兽医学报 2021年12期
袁泽湖,葛 玲,李发弟,乐祥鹏*,孙 伟1,*
(1.扬州大学 教育部农业与农产品安全国际合作联合实验室,扬州 225000;2.兰州大学草地农业科技学院 草地农业生态系统国家重点实验室/农业农村部草牧业创新重点实验室/教育部草地农业工程研究中心,兰州 730020;3.扬州大学动物科学与技术学院,扬州 225000)
全基因组选择(genomic selection,GS)[1]利用覆盖全基因组的标记信息计算全基因组育种值(genomic estimated breeding value,GEBV)。相较于传统的育种方法,GS通过对拟留种的个体进行早期选择和增加选择的准确性进而加快育种的遗传进展[2]。通过改进GS方法无法再缩短育种的世代间隔,因而如何提高GS的准确性以获得额外的遗传进展一直是GS研究的核心问题。
全基因组测序(whole genome sequencing,WGS)技术逐渐成熟,测序成本不断降低,对家畜进行大规模测序已成为可能。GS从基于标记与因果突变连锁的GS0.0时代(如利用50K芯片进行GS)发展到基于全基因组变异的GS2.0时代[3]。WGS检测的变异已包含了所有因果突变位点,因此GS2.0不再受标记与因果突变连锁不平衡(linkage disequilibrium,LD)的限制。理论上,基于全基因组变异的GS准确性应高于芯片(如50K)GS的准确性。然而,有研究结果显示当不考虑生物学先验信息时,基于全基因组变异的GS准确性并不比基于芯片的GS准确性高[4-6]。当前,各种组学技术不断成熟,从公开的资料或前期的研究积累获取生物学先验信息已比较容易。因而,如何在GS模型中整合已知的先验信息,进而通过提高GS的准确性获得额外的遗传进展成为当前动物育种研究的一个重要的课题。本文首先对生物学先验信息的类型以及整合先验信息的GS方法进行综述,并探讨了这些方法在家畜育种中的应用和前景,以期为家畜开展整合生物学先验信息的GS研究提供借鉴与参考。
1 生物学先验信息与先验信息获取群体
1.1 生物学先验信息
生物学先验信息,即预先知道的遗传学知识,由于单核苷酸多态性(single nucleotide polymorphisms,SNP)是育种中广泛使用的分子标记,因而对于GS来说,寻找生物学先验信息的核心任务是根据已知的遗传学知识对SNP的遗传贡献(即对重要经济性状的影响程度)进行排序或者分类。理论上,任何能够影响表型的各组学数据或已知的遗传学知识都能作为GS的生物学先验信息,因而先验信息可包括基因组、表观组、转录组、蛋白组、代谢组(图1A)以及已知的基因功能注释或SNP注释等信息(图1B)。
在基因组层面,全基因组关联分析(genome-wide association study,GWAS)的结果是GS最常用的一种生物学先验信息[7-21](表1),根据GWAS的P值和(或)标记效应很容易获得SNP对某一性状遗传贡献大小的信息(P值越显著的遗传贡献越大,标记效应大的遗传贡献大)。除GWAS外,选择信号的分析结果的也能作为GS的先验信息,如根据群体分化指数(FST)获得SNP遗传贡献的大小[22-23]。转录组数据也是一种重要的生物学先验信息(表1),位于差异表达基因(differentially expressed genes,DEGs)内的SNP位点通常比其它SNP遗传贡献大[14]。同样,表观组的数据,如组蛋白修饰信息可作为GS的先验信息,因为位于组蛋白修饰区域的SNP比其它SNP具有更大的遗传贡献[24]。当前,蛋白组和代谢组数据还少有用作GS先验信息的报道[25]。此外,数据库的注释信息也是一类重要的先验信息,如基因本体(gene ontology,GO)注释信息[7,26-27]、京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路注释[26]、动物数量性状基因座数据库(AnimalQTLdb)[28-30]等。整合不同类型的生物学先验信息,GS准确性的增幅并不相同[8],可见,不同的组学数据能够提供不同先验信息。目前的研究主要利用单一维度的先验信息,随着多组学数据的积累,如何系统地整合多组学数据并给予SNP恰当的遗传贡献仍面临挑战。最近有学者做了一些尝试,Xiang等[25]在奶牛的研究中提出了功能与进化性状遗传力打分方法(functional-and-evolutionary trait heritability,FAETH),FAETH根据SNP的遗传力对标记的遗传贡献进行排序,并在奶牛的GS中取得了较好的应用效果[31]。然而,计算FAETH需要多次计算SNP的遗传力,计算量大;在将来,应开发更多的算法整合多组学数据对SNP的遗传贡献进行排序或者分类。
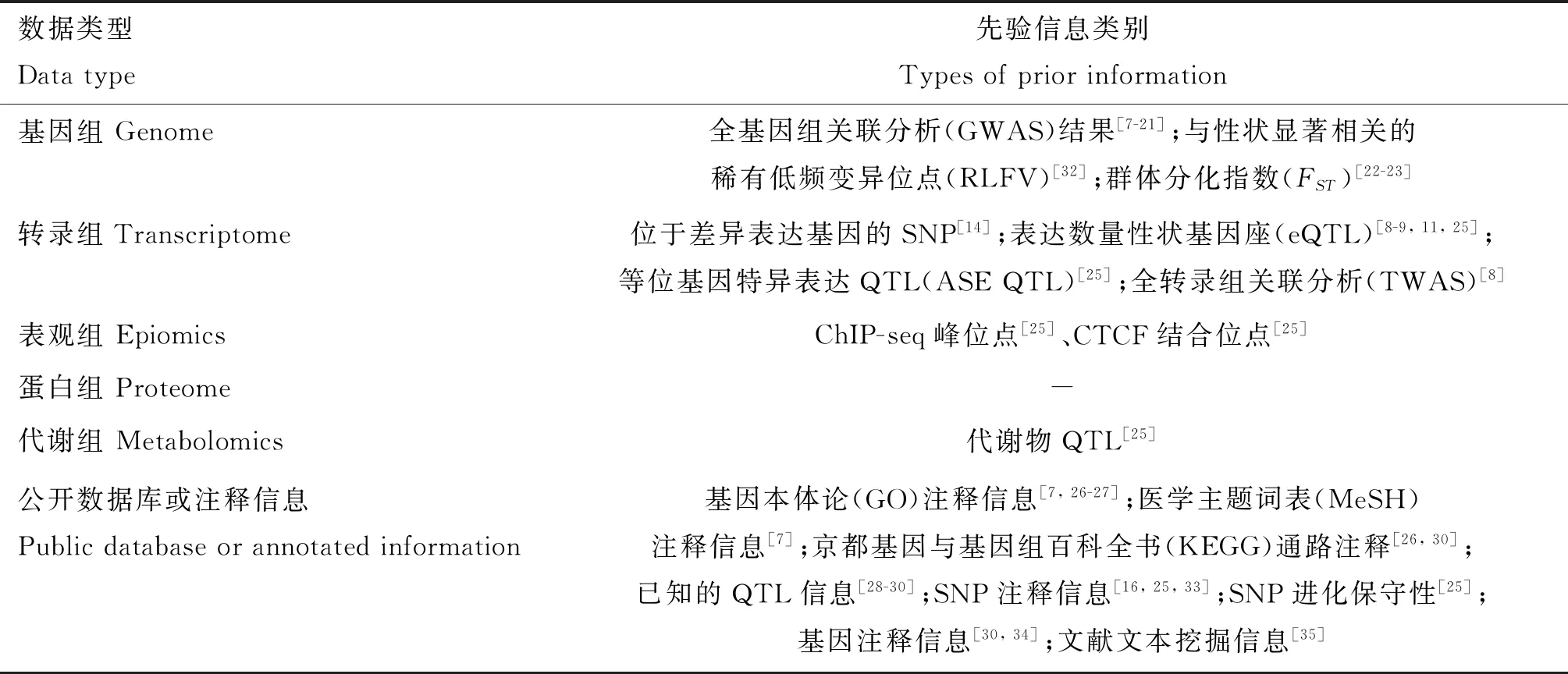
表1 生物学先验信息类别Table 1 Type of biological prior information
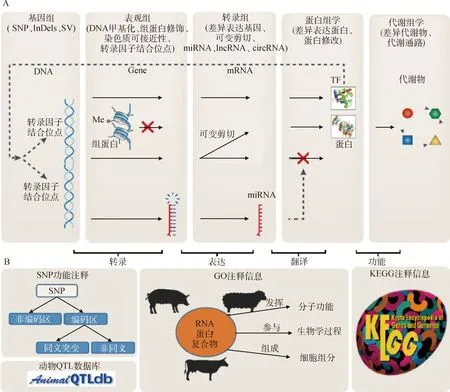
A.多组学数据;B.注释信息A.Multi-omics data;B.Annotation information图1 潜在的生物学先验信息Fig.1 Potential biological prior information
1.2 先验信息获取群体
在早期整合生物学先验信息的GS研究中,由于可获得的先验信息相对匮乏,因而先验信息通常来自参考群体,比如,先在参考群体进行GWAS后[18,36],再将GWAS的结果作为GS的生物学先验信息。此外,有学者为了提高计算效率和降低分型成本,仅使用先验信息(如GWAS显著的SNP位点)进行GS[9,13-14]。这些尝试为生物学先验信息的利用奠定了重要基础。然而,先验信息获取群体与参考群体使用相同的数据和(或)仅使用先验信息进行GS时导致GS偏差(bias)变大[9,18,36]。偏差较大的原因可能是只利用部分标记无法准确地捕获群体的遗传结构信息或人为有选择地使用了随机的遗传信息[13]。因而,Macleod等[14]提出整合生物学先验信息的GS研究需要专门的先验信息获取群体,且要求先验群体与参考群体和验证群体相对独立;此外,不能仅使用先验信息而需要在基准芯片(如50K)的基础上添加先验信息,进而减少GS的偏差。随后,在绵羊部分肉用性状中按照这个方式划分各类群体进行GS,结果发现在50K芯片的基础上整合GWAS和eQTL先验信息并没有明显改变GS的偏差[11]。在实际应用中,应根据先验信息的类型决定是否需要先验信息发现群体,当利用数据库注释信息时,不需要先验信息获取群体。
2 整合生物学先验信息的全基因组选择方法
除了获得生物学先验信息以外,另一个核心的问题是如何应用这些先验信息,即开发合适的GS方法。当前整合生物学先验的GS方法大多是在GBLUP或者Bayes方法的基础上改进而来。整合生物学先验信息的GS方法按照整合先验的方式,可以大致分为3大类:第一类是根据生物学先验信息给予不同的SNP以不同的遗传权重,简称为位点特异的方法;第二类是根据生物学先验信息将SNP划分为几个不同的类别,给予不同类别的SNP以不同的遗传权重,简称类别特异的方法;第三类是将效应较大的SNP位点当作协变量,简称为协变量方法。
2.1 位点特异的方法


表2 G矩阵加权系数汇总信息Table 2 Summariy information of weighted index (di)for G matrix
加权系数di主要来源于GWAS的P值,标记效应或标记方差。Gianola等[41]指出,加权系数di要能反应标记所能解释的遗传方差。Su等[18]在奶牛奶用性状中的研究结果显示,利用后验标记方差对G矩阵进行加权优于基于P值和标记效应的平方。Ren等[40]通过比较几种不同的加权策略发现不同的加权策略适合不同性状。因而,不同的性状需根据其遗传结构选择适合的加权系数。
获得加权系数di需要预先计算出每个标记的效应大小及加性遗传方差,因而计算量非常大。Zhang等提出了BLUP|GA方法[42-43],该方法通过构建T矩阵来替代G矩阵:
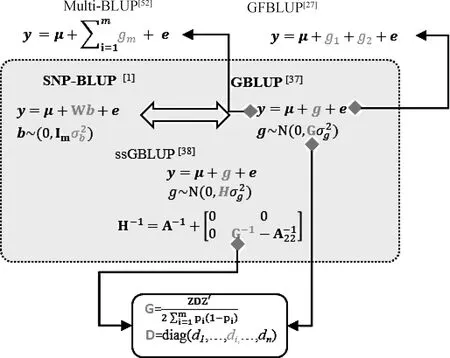
虚线框之内的方法不能整合生物学先验信息;虚线框之外的方法能够整合生物学先验信息;箭头表示方法的演变过程;公式中y为表型向量;μ为群体均值向量,b为标记效应,其效应服从正态分布N(0,Imσ2b),Im为单位矩阵,σ2b为标记效应方差。W为基因型矩阵;e为随机残差;g为基因组估计育种值向量,其值服从正态分布N(0,Gσ2g),G为基因组亲缘关系矩阵,σ2g为加性遗传方差;A为基于系谱的亲缘关系矩阵;H为一步法GBLUP的亲缘关系矩阵;Z为校正后的基因型矩阵;D为亲缘关系的加权对角矩阵,对角线元素为diThe methods within dashed box cannot integrate biological prior information;the methods outside the dashed box can integrate biological prior information;arrows indicate the evolution of the methods;y is a vector of phenotype;μ is a vector of population mean value;b is marker effects following a normal distribution N(0,Imσ2b),where Im is an identity matrix,σ2b is the variance of marker effect;W is a matrix of genotype;e is a vector of random effect;g is a vector of genomic estimated breeding value following a normal distribution N(0,Gσ2g),where G is a genomic relationship matrix,σ2g is the additive genetic variance;A is a pedigree-based relationship matrix;H is a relationship matrix for ssGBLUP;Z is a matrix of scaled genotype;D is a diagonal matrix of weighted index for G matrix,where di is the ith element in main diagonal图2 基于最佳线性无偏估计(BLUP)的全基因组选择方法Fig.2 Genomic selection methods based on best linear unbiases prediction (BLUP)

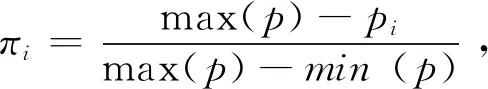

虚线框之内的方法不能整合生物学先验信息;虚线框之外的方法能够整合生物学先验信息;箭头表示方法的演变过程;公式中ui为标记效应,在不同的方法中服从不同的分布;π为效应值为零的标记的比率,在不同的方法中值不同。P表示关联分析P值The methods within dashed box cannot integrate biological prior information;the methods outside the dashed box can integrate biological prior information;arrows indicate the evolution of the methods;ui is the marker effect which follows different distribution in different methods;π is the ratio of marker with effect equal to zero;P is the P value of association analysis图3 全基因组选择的Bayes方法Fig.3 Genomic selection methods based on Bayes
2.2 类别特异的方法
2.2.1 基于BLUP的类别特异方法 有学者提出了另一类整合先验的GFBLUP方法(图2),与之前的对每一个SNP赋予不同的遗传权重不同,GFBLUP方法根据SNP位点的“遗传贡献”将其分为不同的类别,并通过拟合两个及以上的G矩阵赋予不同类别的SNP以不同的遗传权重[27,50],即:
y=1μ+g1+g2+e
式中,参数的含义与GBLUP相同,g1表示第一组标记的GEBV,g2表示第二组标记的GEBV。与此同时,有学者提出了与GFBLUP类似的MultiBLUP方法[51],即:
式中,参数的含义与GFBLUP类似。GFBLUP和MultiBLUP都能拟合两个及以上的随机变量。当样本数较大或者标记的密度过高时(如全基因组测序鉴定到的SNP),由于计算资源或者软件本身的限制,拟合两个或多个G矩阵的GFBLUP和MultiBLUP方法大大增加了计算负担。为了减少计算资源,两个或多个G矩阵还可以通过加权的方法进一步简化:
y=1μ+gTotal+e

2.2.2 基于Bayes的类别特异方法 在大多数情况下,BayesR的准确性优于其它的Bayes方法,近年来有学者开发了简化版的BayesR方法SBayesR。SBayesR将BayesR模型拓展到了概述统计数据并通过考虑SNP之间的连锁不平衡(linkage disequilibrium,LD)信息将稠密亲缘关系矩阵变为稀疏矩阵以提高计算效率[52]。目前,类别特异的Bayes方法大多是从BayesR的基础上改进而来的。Brøndum等[53]提出了BayesRS方法,这一方法的思想与mutiBLUP类似,都是根据SNP集的方差组分,赋予不同类别的SNP以权重的先验信息。Macleod等[14]在BayesR的基础上提出了BayesRC方法,BayesRC与BayesR之间的主要区别在于BayesRC允许不同类别的SNP独立的计算标记效应来整合生物学先验信息[14],BayesRC和BayesRS主要的区别在于BayesRS允许不同的SNP集有不同的先验信息而BayesRC则允许不同的SNP集有不同的后验效应(图4)。
2.3 协变量方法
与之前的方法不同,有学者直接提出了将效应大的位点作为协变量(covariate)放入混合线性模型中以提高GS的准确性[16,54]。在奶牛繁殖性状(sire conception rate,SCR),通过这种方式将GS的准确性从0.340增加到0.403[16]。协变量方法适用于已鉴定到因果突变的性状;然而,家畜大多重要经济性状受多个QTL影响,且大多性状的因果突变位点仍未解析,当因果突变未知时,采用这种方法可能会因为加入了错误的协变量而降低GS的准确性,因而这种策略无法大面积提高GS的准确性。
2.4 基于机器学习策略的协变量及位点特异整合方法
最近,我国学者提出了KAML(Kinship Adjusted Multiple Loci Best Linear Unbiased Prediction)方法[55]。该方法主要通过两个途径提高GS的准确性,一是通过将效应大的位点当做协变量加入线性模型中;二是对G矩阵进行加权,加权系数为:
其中,α为对数函数的基数(待估),β是加权的比率(待估),P为GWAS的P值,m为标记数目。与之前整合先验信息的方法不同,KAML通过机器学习方法(交叉验证、多元回归、网格搜索以及二分求极值等)智能化地选择将哪些位点当作协变量以及计算加权系数的值(即估计α和β)而非人为预先指定固定的值。一方面,KAML通过机器学习算法整合各种方法的优点,因此其准确性较高,目前已接近Bayes方法[55];另一方面,KAML也难以完全消除已有方法的缺陷,例如,利用单一的组学数据筛选效应较大的位点难免会出现假阳性。
3 整合生物学先验信息的GS在动物育种中的应用
3.1 整合生物学先验信息的GS在牛育种中的应用
产奶性状是奶牛最重要的一类性状,在奶牛中,品种内整合生物学先验信息的GS方法准确性增幅较低,如,Su等[18]利用位点特异的GBLUP方法,在奶产量、乳脂产量、乳蛋白产量等性状的GS中提高了2%的准确性。Macleod等[14]利用BayesRC方法在奶产量、乳脂产量、乳蛋白产量的GS提高了准确性,但平均增幅度较小(<1%)。Mouresan等[56]整合QTL先验信息,在奶产量、乳脂率的GS中分别提高了0.014和0.051的准确性。Liu等[17]在产奶性状中,利用SNP注释先验信息,并没有提高GS的准确性。然而,品种间整合生物学先验信息的GS方法的准确性增幅远高于品种内的GS。Fang等[26,57]整合GO、KEGG、差异表达基因等信息,发现跨品种的GS准确性幅度远大于品种内的增幅。在牛奶脂肪酸相关性状的GS中也有类似发现,整合GWAS结果在荷兰奶牛群体中平均提高38%的准确性,在丹麦群体中平均提高23%,在中国群体中平均提高13%[10]。可见,整合生物学先验信息进行GS时,亲缘关系的远近是影响准确性的一个重要因素。当亲缘关系比较近时(如品种内),不同分子标记之间处于高度连锁不平衡状态,很难将先验信息准确地赋予给相应的分子标记[58]。因而对于奶牛的育种,合理利用生物学知识,将有可能提高品种间(或亲缘关系较远的群体)的基因组预测准确性[59]。然而,不同品种之间QTL的效应的大小和方向、最小等位基因频率可能不同,使得品种间的预测变得更加复杂[58]。
在Hanwoo肉牛的研究中,通过整合基于文本挖掘的先验信息利用GFBLUP方法,提高了背膘厚、眼肌面积、半膜肌剪切力、背最长肌剪切力、半膜肌肌内脂肪含量和背最长肌肌内脂肪含量GS的准确性[35]。在另一个研究中,整合GWAS和eQTL的先验信息利用加权GBLUP和BayesR对Hanwoon牛胴体重、大理石花纹打分、眼肌面积、背膘厚进行GS,准确性提高了0.01~0.05[9]。随后,Mehrban等[60]通过加权ssBLUP,发现Hanwoon牛胴体性状GS和周岁重GS的准确性分别提高了71%和99%。国内肉牛整合生物学先验信息进行GS也取得了重要进展,Xu等[33]通过整合SNP的注释信息,采用方法多种策略对胴体重、宰前活重以及上脑重进行GS,结果发现准确性提高5.4%~9.8%。肉牛育种可以通过整合生物学先验信息的GS进行早期选育以及通过整合生物学先验信息提高GS的准确性以提高遗传进展。
3.2 整合生物学先验信息的GS在猪育种中的应用
与奶牛育种不同,猪的GS很难通过早期选育缩短其世代间隔[61]。因此,在猪的育种中,通过整合生物学先验信息提高GS的准确性,尤其是低遗传力的性状[62]以获得额外的遗传进展十分有意义。传统的GS方法对猪的生长或胴体性状已具有很好的预测准确性[61],当整合适当的先验信息和选择合适的GS方法,还能再提高眼肌深度[63]和日增重[63]、瘦肉率[28]等性状的准确性。然而,传统的GS对母猪的繁殖性状进行选择,准确性非常有限,因为母猪的参考群体较小且遗传力较低[61]。有研究发现,整合生物学先验信息的GS方法具有进一步提高提高母猪繁殖性状准确性的潜力[29]。若通过整合人、小鼠以及其它物种繁殖研究的信息以及猪繁殖性状相关基因的功能注释信息,整合生物学先验的GS方法在母猪的繁殖性状中具有较大的应用潜力。
3.3 整合生物学先验信息的GS在羊育种中的应用
相较于牛和猪,羊的产值低、遗传多样性高、经济性状种类多,包含肉用、毛用、奶用、繁殖等性状。因而,开发高精度、低成本、多性状的GS方法才能支撑羊的高效育种。在50K芯片的基础上,通过整和基于重测序的GWAS先验信息、eQTL的先验信息均能提高绵羊重要经济性状GS的准确性[13]。可见,在50K基准芯片的基础上优化GS的方法和使用先验信息可提高GS的准确性,且不至于额外增加过多的经济和计算负担。在低密度芯片的基础上通过整合生物学先验信息以提高GS的准确性,可能是以后羊GS育种的可行之策。
4 存在问题与展望
GS的准确性与遗传进展成正相关,与传统的GS方法相比,整合生物学先验信息的方法通过提高GS的准确性以获得额外的遗传进展。当前的生物学先验信息大多来自单一组学的数据,所能提供的信息有限,随着家畜基因组功能注释信息的持续积累和完善[64],如何系统地整合多组学的数据并准确地给予分子标记以准确的遗传权重仍需要开发新的方法。此外,当前整合生物学先验信息的GS方法都有各自的缺陷,位点特异的方法需要获得每个标记的遗传贡献,因而能整合的先验信息类型有限;类别特异的方法只是粗略地将分子标记划分为多个类别,划分的方式往往具有很大的主观性,并没有严格的科学依据,且每个类别内标记的遗传贡献仍然是相同的,不符合科学假设;协变量方法需要准确获得效应较大的位点,但大多经济性状是由微效多位点所控制的,因而其适用性有限。因此,将来应该开发出计算速度快、准确性高、能整合所有类型生物学先验信息且适用所有性状的GS方法。当前国内外的研究主要是将多组学数据作为GS的先验信息以提高GS的准确性,而不是将其直接整合到GS中。随着测序技术的发展和测序成本的降低,将来也可能获得大规模的多组学数据。在将来,可能在主流的GS方法BLUP、Bayes、机器学习、深度学习等的基础上开发出可以直接整合多组学数据的GS方法。可以预见,随着生物学先验信息越来越精准,适用范围广、准确性高、速度快的GS方法被开发,整合生物学先验信息的GS将会在家畜育种中发挥重要作用。
猜你喜欢
现代畜牧科技(2021年4期)2021-07-21
建材发展导向(2021年10期)2021-07-16
成都信息工程大学学报(2019年3期)2019-09-25
福建基础教育研究(2019年1期)2019-09-10
福建基础教育研究(2019年1期)2019-05-28
自动化学报(2017年5期)2017-05-14
光学精密工程(2016年4期)2016-11-07
电子制作(2016年21期)2016-05-17
管理现代化(2016年5期)2016-01-23
探测与控制学报(2015年4期)2015-12-15
