
基于全转录组的心力衰竭预测标志物
2021-12-29 00:51李丹丹张常建陈伟浩乔媛媛
转化医学杂志 2021年6期
马 伟,李丹丹,张常建,陈伟浩,熊 鸣,乔媛媛,李 军
心力衰竭(heart failure,HF)是一个复杂的临床症状,其特点是心脏供血不足[1]。在发展中国家,HF 是患者住院的常见原因,其发病率和病死率也高[2-5]。HF 的发病率逐年增加,已经成为重要公共健康问题[6-8]。在发展中国家,HF非常普遍,其患病率高达0.4%~2%[9-10]。虽然现在的诊断和治疗水平已经改善了HF 患者的预后,但是其5 年生存率仍不足50%[11-12]。HF 越早发现,患者预后越好,因此寻找预测HF的标志物对于降低患者病死率以及提高患者生活质量非常有帮助。目前也发现了一些HF 的风险因子。例如,Onoue 等[13]发现肌肉减少症可以预测HF 患者的不良事件,而肌肉减少症可以通过结合年龄、握力和小腿围来进行评估。Soyama 等[14]通过超声心动图监测心脏发现心脏舒张应变(diastolic wall strain,DWS)能够评估HF 患者心脏的功能。他们发现低DWS 水平患者,心功能比较好,而左心室功能受损的患者DWS 值升高,DWS 高的患者预后往往不好。近些年来,随着高通量分子实验技术的发展,多种多样的基于血液或组织的分子标志物也出现了。Huang等[15]发现外周血白细胞的线粒体DNA(mitochondrial DNA,mtD‐NA)的拷贝数变异(copy number variation,CNV)是HF 的独立危险因素,能够预测HF 患者病死率。Tumorigenicity-2 (sST2)能够为HF 患者的后期处置提供帮助[16]。Circulating cell-free DNA (cfDNA)也可以作为HF 标志物[17]。在失代偿期的HF 患者,血清高水平内源性促红细胞生成素的长期的临床预后更差[18]。HF 标志物的概况参见这三篇综述[19-21]。芯片技术的发展和二代测序及三代测序的发展,累积了很多全转录组数据,这也为开发新的多分子标志物带来了机遇和挑战[22-23]。通过转录组分析,Maciejak 发现RNAS1、FMN1和JDP2与HF 患者的临床指标有显著的相关性。ST 段太高的心梗第一天,这三个基因的表达值在随后发生HF 的患者中高于随后未发生HF 的患者[24]。基于转录组的方法能够发现基因表达特征作为疾病的标志物[25]。Heidecker基于转录组发现了一个45个基因特征的标志物,可以评估HF 患者的风险[26]。然而上述标志物部分是单个分子,部分是几十个基因的特征,相比人类将近3 万个基因的数量,标志物涉及到的基因数量还是特别少,这就会导致容易产生过拟合,使得标志物的扩展应用性不强。因此非常有必要寻找基于转录组的扩展性好的标志物。
使用全转录组的特征而不是仅仅局限于差异基因可以产生更稳定的HF标志物。传统的观点认为蛋白质功能的改变是由于突变导致了氨基酸序列发生了变化,然而有人发现原子含量,尤其是氧原子也与蛋白质功能有关。大气中氧含量的增加促进细胞交流与细胞器的形成,跨膜蛋白长度与蛋白质氧含量有关,而蛋白质氧含量又受到大气氧含量的约束[27]。与无氧呼吸比较,有氧呼吸能够有效的获取能量,发生更多的代谢反应[28-29]。对于HF患者,其特征就是组织缺氧,细胞氧化应激增加[30]。因此,我们猜想在这样的缺氧情况下,转录组会产生系统性的缺氧反应,不同氧含量的蛋白质其表达量会有不同的变化,这种不同的变化可以用来寻找HF 标志物。为了证实此猜想,本课题组从GEO(Gene Expression Omnibus,GEO)下载了两套HF 数据(GSE57345 和GSE21610),然后开发了一个基于全转录组的预测HF的标志物[25,31],发现基因的表达水平与氧原子含量(oxygen atom content,OAC)的Spearmam’s相关系数能够很好的区分HF。
1 数据与方法
1.1 蛋白质、mRNA 序列与基因表达数据 本研究从Ensembl 数据库下载了蛋白质序列和mRNA 序列[32]。从GEO 数据库下载了两套HF 数据,筛选标准为:组织来源为心肌组织活检并且有正常心肌组织,最终筛选获得GSE57345(Affymetrix Human Gene 1.1 ST Array)和GSE21610(Affymetrix Human Genome U133 Plus 2.0 Array)[33]。在GSE57345 中,心脏组织样本是MAGNet 联盟从患者取得。本文的数据分析采用R 语言进行统计分析(version 3.6.2,https://www.r-project.org/)。
1.2 计算蛋白质氧原子含量以及OAC 与基因表达的Spearman 相关性 蛋白质氧原子含量定义为:蛋白质序列中氧原子的数量/蛋白质总原子数。在计算相关性之前,先根据转录本最优密码子的比例(codon optimality ratio of protein,CORP)进行排序,然后分成10 等份。CORP 定义为:mRNA 中最优密码子数量/总的密码子数量。最优密码子定义为:密码子的最佳稳定性相关系数得分(correlation coefficient,CSC),而正值得分的密码子就是最优密码子[34]。首先计算所有的蛋白质编码转录本的表达量与其相应蛋白质OAC 的Spearman 相关系数,然后针对上述10 份的每一份再计算一次。因此,对于每个样本,会得到11 个相关系数,1 个总的和10个10等份的。
1.3 构建分类器 使用randomForest R包构建随机森林模型,模型参数为:树的数量为800,其他参数为默认值[35]。在构建分类器这里,将两套数据的相关系数范围使用公式(1)缩放至[-1,1],y’表示放缩后的值,y表示放缩前的值,min 表示原数据值中最小值,max 表示原数据中最大值。10 个10 等份的相关系数作为输入特征。然后通过特征重要性筛选特征。使用准确率、阳性预测值(positive pre‐dictive value,PPV)和阴性预测值(negative predic‐tive value,NPV)作为分类器性能评估参数。使用5折交叉验证、测试集以及独立验证集测试模型准确性。准确率=(真阳性数量+真阴性数量)/总量;PPV=真阳性数量/(真阳性数量+假阳性数量);NPV=真阴性数量/(真阴性数量+假阴性数量)。
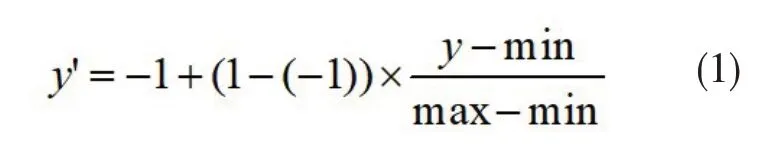
2 结果
2.1 数据统计学特征 数据的人口统计学资料和基本临床信息见表1。GSE57345 包含313 个样本,包括177 个HF 样本和136 个正常样本(non-HF con‐torls,NF)。HF样本中有男性144例和女性33例,正常样本有男性73例和女性63例。两组的平均年龄分别为55.4 和49.4。GSE21610 有38 个样本,30(28例男性和2例女性)和HF样本和8个正常样本(6例男性和2例女性),两组平均年龄分别为51.3和29.0。
2.2 HF患者OAC与基因表达的相关性 首先对所有编码蛋白的转录本计算了OAC 与基因表达的相关性。发现在GSE57345数据集中,HF 样本的相关系数远高于正常样本(图1A,P=2.25×10-11)。然后再使用相关系数作为参数做受试者工作曲线(re‐ceiver operating characteristic curve,ROC),曲线下面积(the area under curve,AUC)为0.772,如图1B 所示。数据集GSE21610的结果类似(P=0.04),AUC为0.729,见图1C和1D。数据集GSE21610没有数据集GSE57345 显著可能是由于其样本量的原因(38 VS 313)。虽然两个数据集中的相关系数差别比较大,但是HF样本都高于正常样本。
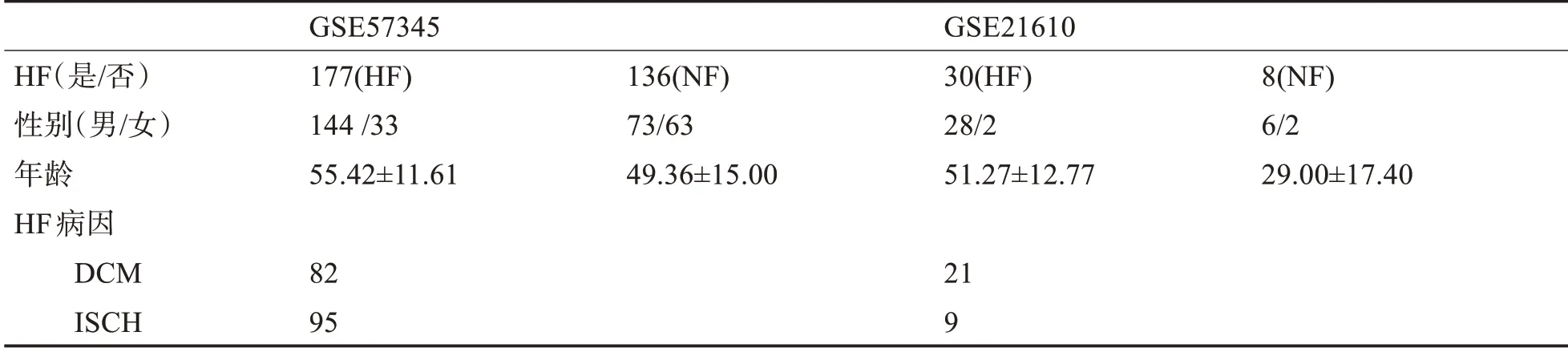
表1 数据集基本信息

图1 两个数据集中所有转录本的相关系数
2.3 基于选择特征的随机森林分类器 转录本最优密码子的比例与转录本的翻译速度有关系[34]。因此,我们根据转录本的COPR 将所有转录本等分为10 组,每组分别计算表达水平与氧含量的Spearman 相关系数。在数据集GSE57345 中,第3、5、6、7、8、9、10 部分有显著的区分能力,AUC 大于0.6,表2。尤其是第5 部分和第8 部分,AUC 大于0.8。第4 部分的AUC 也将近0.6(0.599)。在所有10部分中,HF样本的相关系数都大于正常样本。

表2 总体和10部分相关系数的区分能力
为了构建分类器,本研究将GSE57345 随机分为训练集和测试集两部分,训练集包含250 个样本(142 HF 和108 NF),测试集包含63 个样本(35 HF和28NF)。 为了评估分类器的性能,把数据集GSE21610 作为完全独立的一个测试集,仅仅在构建好分类器后才使用。为了保持数据的通用性,再将两套数据的相关系数范围都缩放至[-1,1]。然后使用训练集进行模型构建。10 部分的相关系数作为特征输入随机森林模型,根据特征重要性得分,选择了前三个重要性得分最高的特征(图2A),分别为第3、5、8部分。最终使用这三个特征作为输入特征构建随机森林分类器。首先,使用5 折交叉验证测试分类器性能。5 折交叉验证的AUC 为0.851(图2B),其准确率为83.6%,PPV 为85.7%,NPV 为81.5%,结果虽然不错,但是也可能是由于过拟合导致,必须通过测试集进行验证。使用训练集所有样本构建模型,然后使用GSE57345 的测试集部分数据进行测试,其AUC 为0.793(图2C),准确率、PPV和NPV 分别为77.8%、80.0% 和75.0%。最终,在完全一个独立的数据集GSE21610上测试了我们的分类器(图2D),AUC高达0.908。准确率、PPV和NPV分别为86.8%、86.7% 和87.5%。总体上,本研究构建的分类器能够很好地区分HF和NF(表3)。

表3 分类器的性能评估
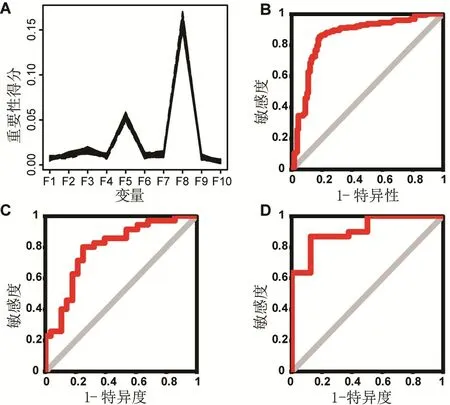
图2 随机森林模型性能评估
在独立的测试集GSE21610中,HF和NF的年龄不匹配。为了排除年龄的影响,我们挑选了10个年龄在11.6 至46.4(NF 样本年龄的)的HF 样本来匹配NF样本。这18个样本(10个HF 和8个NF)的准确率、PPV和NPV分别为83.3%、80.0%和87.5%。这说明我们的分类器在独立测试集上的效果不受年龄影响,也并非正负样本差引起。
3 讨论
在本研究中,本研究发现转录本表达量与转录本对应蛋白质的氧原子含量的相关性能够很好地区分HF 与NF。相关性在HF 组中更高,这说明在缺氧环境下,转录组受到了很大影响,系统更倾向于表达氧原子含量更高的蛋白质,可能是为了补偿缺氧或者是缓解氧化应激压力。通过交叉验证、测试集以及独立测试集评估,发现利用表达量-氧原子含量相关性构建的分类器展现了非常稳定的预测性能。本研究开发的基于全转录组的策略能够避免少量表达波动带来的影响。
然而本研究仍然存在缺陷。首先,受限于数据库中HF 患者心脏组织表达谱数据的限制,仅仅评估了其区分HF 和NF 的性能,没有评估其与HF 患者预后的关系。其次,使用的数据集,其样本来源是心肌组织(作为临床预测标志物,更容易取材或者非侵袭的取材更方便合适,比如血液、尿液等)。现实中,常用的很多HF 标志物确实来自血液或者是身体检查。尽管如此,也有基于心肌组织的标志物。Heidecker 用心肌活检组织找到了HF 的转录组标志物[26]。Morgun 使用心肌活检组织转录组构建了预测是否发生心脏移植排斥反应的预测器,提高了心脏移植排斥诊断率[36]。实际上,心肌活检组织是诊断心脏移植排斥反应的金标准。最后,本研究开发的标志物是基于整个转录组的,不是基于个别基因或者少数基因的,无法对其功能做阐释。我们发现转录效率低的转本部分(F1、F2、F3)对HF鉴别无作用,而转录效率高的部分对HF 有鉴别作用,这可能与人类进化过程中氧含量的变化有关,也可能与人体反应机制有关。在缺血情况下,心肌组织氧含量高的转录本转录更多,可能是反应性的存储更多的氧而导致的。随着技术的发展,心肌组织样本提取会变得越来越容易,侵袭性越来越小,基于心肌组织的标志物会越来越多。由于HF是临床死亡最多原因之一,多变量的标志物将会更好的预测HF,改善其预后[37]。因此,如果有合适的数据,我们非常乐意在新的数据中检验我们预测器预测HF及HF预后的能力。
总的来说,本研究构建了一个基于全转录组的HF 标志物,能够准确的区分HF 与NF。随着全转录组技术的成熟,价格越来越低,本研究提出的策略能够很容易的实施,未来将会有更多的转录组标志物甚至多组学整合策略的标志物出现。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
电子产品世界(2022年4期)2022-04-21
健康体检与管理(2022年2期)2022-04-15
福建农业学报(2021年6期)2021-08-18
天津医科大学学报(2021年2期)2021-03-29
计算机系统应用(2021年2期)2021-02-23
教学考试(高考生物)(2020年4期)2020-11-18
电子技术与软件工程(2019年18期)2019-11-18
发明与创新·中学生(2019年6期)2019-06-26
安徽农业科学(2018年1期)2018-05-14
