
罕见病研究:CTNS基因突变致胱氨酸贮积症
2021-12-22 03:31:12王欣张碧丽陈晓颖郭珍
中国当代儿科杂志 2021年12期
王欣 张碧丽 陈晓颖 郭珍
(天津市儿童医院特需病房,天津 300134)
胱氨酸贮积症(cystinosis)是一种常染色体隐性遗传病,临床较为罕见,可累及全身各个系统。其发病原因与CTNS基因突变相关,由于该基因突变可导致溶酶体膜上缺乏L-胱氨酸转运蛋白(cystinosin),溶酶体内的胱氨酸无法正常转运,大量蓄积后引起眼、肾脏、神经、内分泌腺等多器官功能障碍[1]。目前胱氨酸贮积症的主要治疗方法包含对范科尼综合征及肾外并发症的对症治疗及应用半胱胺药物特异性清除胱氨酸[2]。通过早期诊断及尽早开始针对性的治疗可以保障患儿有更好的生长发育过程,并且能够延缓终末期肾衰竭及防止其他肾外并发症的发生[3-4]。该病国外报道 发 病 率 为 1/192 000~1/115 000[3], 最 常 见 的CTNS基因突变为 57 kb 碱基缺失[5]。我国仅见个例报道[6-9],CTNS基因突变包含1例IVS8-1(delGT)纯合缺失[6],1例c.696C>G[7],2例c.969C>G[8]及1 例c.681G>A[9]个案报道。该文对天津市儿童医院收治的 1 例CTNS基因突变 c.922G>A(p.Gly308 Arg)致胱氨酸贮积症患儿的临床资料进行回顾分析,复习相关文献资料,提高临床医师对该病的认识。现报告如下。
1 病例介绍
病史:患儿,男,1 岁6 月龄,因发现尿糖阳性1年余,多饮、多尿半年入院。患儿4月龄时因发热行尿常规检查发现尿糖阳性,此后监测尿糖波动在-~4+,血糖正常,平时尿色黄、清,无浮肿。患儿1岁时出现多饮、多尿,日饮水量1 000~1 500 mL,夜间饮水500~1 000 mL,夜间排尿8~9次,尿量约50 mL/次。入院前2个月开始出现食欲欠佳,体重不增。入院前5 d为明确尿糖原因于外院检查时发现血钾2.6 mmol/L,血钠124 mmol/L,院外予口服枸橼酸钾治疗,为进一步诊治来我院就诊。患儿自发病以来,精神反应可,食欲欠佳,大便正常。
既往史、出生史及家族史:患儿系第2 胎第2产,孕39+1周顺产,父母及8 岁哥哥身体健康,否认家族遗传病史。
入院体检:身高74 cm(-2 SD),体重8.2 kg(-2 SD)。智力正常,营养欠佳,体型消瘦,皮下脂肪菲薄,皮肤弹性尚可,无浮肿。双肺呼吸音粗,心音有力,律齐,各瓣膜区未闻及杂音。腹平软,肝脾未触及。四肢活动自如,肌力及肌张力正常。
实验室检查:尿常规示比重1.000(参考值:1.003~1.030),尿蛋白±(参考值:阴性),葡萄糖+(参考值:阴性)。血常规示血红蛋白91 g/L(参考值:110~160 g/L),红细胞压积26%(参考值:36%~50%),红细胞平均体积75.6 fL(参考值:86~100 fL),白细胞13.89×109/L(参考值:4×109/L~10×109/L),单核细胞比率9%(参考值:3%~8%),血小板342×109/L(参考值:100×109/L~300×109/L)。毛森试验示总入量10 000 mL/m2(参考值<3 000 mL/m2),总尿量9 250 mL/m2(参考值<3 000 mL/m2),最高尿比重1.006,最低尿比重1.002,最高尿比重-最低尿比重=0.004(参考值>0.009)。电解质示钠126 mmol/L(参考值:137~147 mmol/L), 钾 3.93 mmol/L ( 参 考 值 : 3.5~5.3 mmol/L), 氯 93.6 mmol/L ( 参 考 值 : 99~110 mmol/L), 镁 0.68 mmol/L ( 参 考 值 : 0.7~0.95 mmol/L)。肾功能示尿素11.36 mmol/L(参考值:1.78~6.42 mmol/L),肌酐57 μmol/L(参考值:15~31 μmol/L),碱性磷酸酶 879 U/L (参考值:142~335 U/L)。血肾素>1 000 μIU/mL (参考值:3.11~41.2 μIU/mL),醛固酮>2 000 pg/mL (参考值:30~160 pg/mL),血管紧张素Ⅱ247 pg/mL(参考值:25~60 pg/mL)。血气分析pH 7.291(参考值:7.32~7.42),pCO220.3 mm Hg(参考值:41~45 mm Hg), BE -14.6 mmol/L ( 参 考 值 : -3~3 mmol/L)。24 h尿糖总量13.62 mmol/24 h(参考值<0.15 mg/24 h),尿钙定量 1.41 mg/(kg·d)[参考值<4 mg/(kg·d)]。随机尿蛋白/肌酐=3.142 (参考值<0.1)。钠排泄分数 (fractional excretion of Na,FENa) 0.17 (参考值:0.5~1),钾排泄分数(fractional excretion of kalium,FEK)31.89(参考值<15),肾小管磷重吸收率(tubular reabsorption of phosphate,TRP)63.51(参考值:80~94),肌酐 清 除 率 53.9 mL/(min·1.73 m2) [参 考 值 : 65~80 mL/(min·1.73 m2)]。胰岛素、C 肽、糖化血红蛋白、25 羟维生素D3及肝功能大致正常。头颅、胸部及右手X 线示颅骨骨质未见异常,心肺膈无异常,右手腕骨化核出现迟缓,肋骨、双侧肱骨近端干骺端及右尺桡骨远端干骺端见佝偻病骨改变。腹部及睾丸B 超未见异常。心电图示窦性心动过速,非特异性T波改变(T波双峰),边缘心电图。眼科检查示患儿角膜基质前1/3见大量结晶,裂隙灯下可见均匀金属丝样遍布全角膜的细小反光点,虹膜纹理清晰,晶状体透明,双眼视网膜散瞳后未见结晶样物质沉着。气相色谱质谱联用法遗传代谢病尿筛查示糖尿和氨基酸尿。液相串联质谱法遗传代谢病血筛查示瓜氨酸/精氨酸、苯丙氨酸/酪氨酸、丁酰肉碱/丙酰肉碱增高;谷氨酰胺/瓜氨酸、游离肉碱降低。
征得家长知情同意,分别采集患儿、患儿父母、患儿兄长静脉血2 mL 进行全外显子组测序,结果显示,CTNS基因存在c.922G>A(p.Gly308Arg)纯合变异,父母及兄长均杂合携带(图1)。有文献报道在肾性胱氨酸病患者中检测到该变异[10],相同位置相同氨基酸改变且为复合杂合状态,反式位点的变异是65 kb“欧洲”型缺失,为已知的致病性变异,符合PM3 证据;有功能学研究提示该变异影响胱氨酸在细胞中的转运[11],符合PS3证据;该变异在人群中的频率极低,ESP 数据(http://evs.gs.washington.edu/EVS)、千人数据库(http://browser.1000genomes.org)、 ExAC 数 据 库(http://exac.broadinstitute.org) 及 gnomAD 数 据 库(http://gnomad.broadinstitute.org)均未见收录,符合PM2 证据;生物信息学软件预测这个变异具有致病性,符合PP3证据。因此,依据美国医学遗传学与基因组学学会(America College of Medical Genetics and Genomics, ACMG) 的 变异 解 读 指南[12],该变异判定为可疑致病性变异。
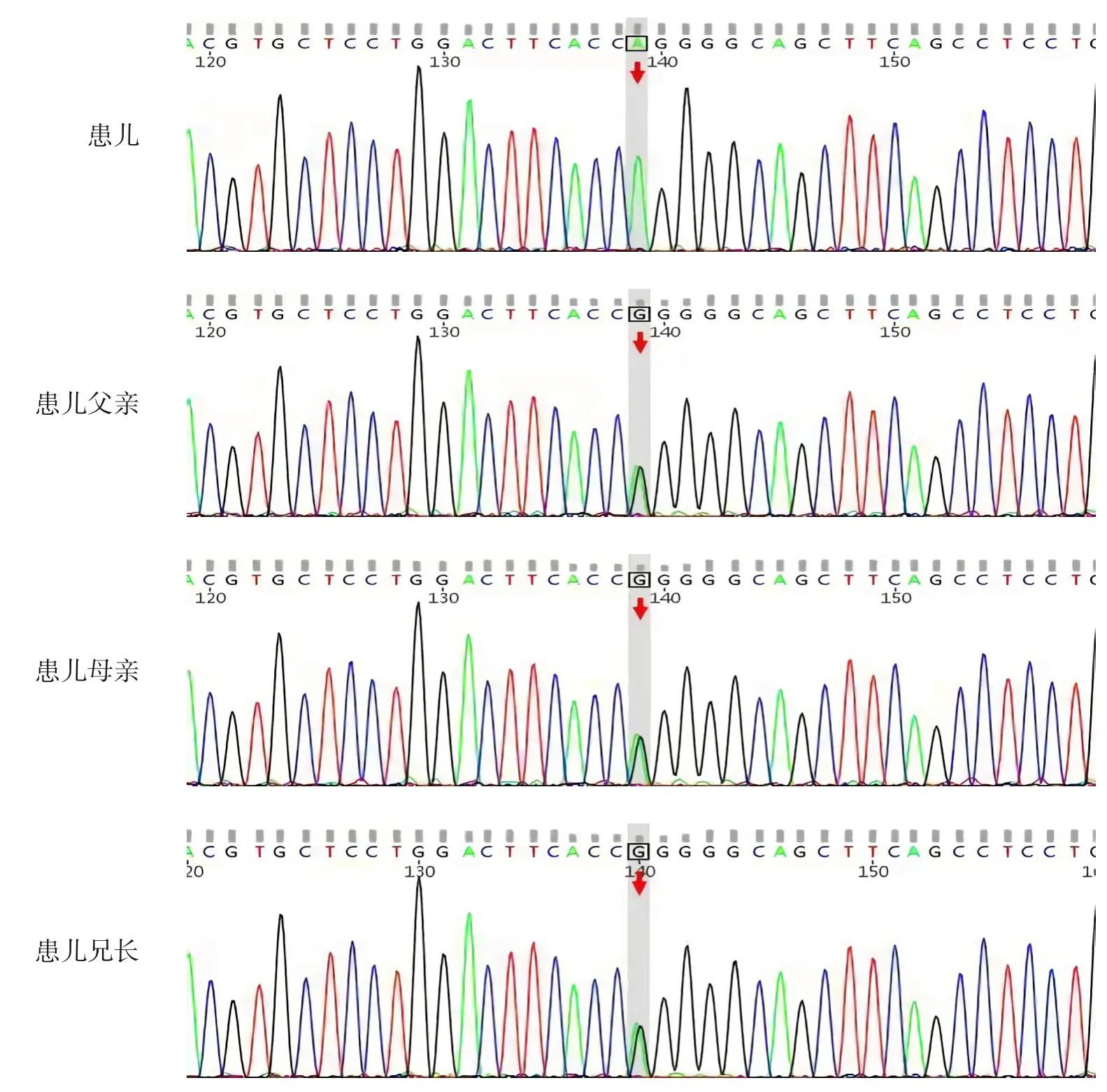
图1 患儿及其父母、兄长的CTNS基因Sanger测序图
2 临床经过
患儿入院后予对症支持治疗,维持内环境稳态,监测血电解质、血气分析,口服枸橼酸钠钾(每日4.8 mL/kg,分4 次口服)补钾、补钠,肌内注射25%硫酸镁注射液(每次0.1 g/kg,隔日1次,共2次)补镁,口服骨化三醇胶丸(上海罗氏制药有限公司,每次0.25 μg,隔日1 次)治疗佝偻病,同时补充左旋肉碱(东北制药总厂,0.5 g/d)。
出院后患儿定期门诊随诊,口服半胱胺酒石酸胶囊(法国,开始每日0.36 g/m2,逐渐增至目前每日1.2 g/m2,分4 次口服)特异性清除胱氨酸。目前患儿2岁3月龄,身高78.5 cm(-3 SD),体重8.9 kg(-3 SD),身高体重无明显改善,考虑与胱氨酸贮积症患儿自身生长发育落后及口服半胱胺治疗初期患儿恶心、呕吐等胃肠道反应明显,影响进食相关。目前监测生长曲线,必要时可应用生长激素治疗。尿量较前略有减少,多次尿常规示 pH 6.5~8,比重 1.003~1.010,尿蛋白-~+,尿糖-~+;多次血气分析示pH 7.384~7.437,BE-3.8~0.1 mmol/L; 多 次 血 电 解 质 : 钠 127~135 mmol/L, 钾 3.53~4.75 mmol/L, 氯 88.1~99.7 mmol/L,镁0.61~0.8 mmol/L;肝肾功能大致正常。
3 鉴别诊断
患儿4月龄发病,尿糖为首发症状,同时伴有多饮、多尿、生长迟缓、贫血、低钾血症、低钠血症、低镁血症、代谢性酸中毒、氨基酸尿、FEK增高、TRP 降低,多部位X 线示肾性佝偻病改变。根据以上临床特点分析,考虑如下相关疾病:(1)糖尿病:患儿长期尿糖阳性,伴有多饮、多尿症状,首先临床需鉴别糖尿病。本例患儿发病后多次检测血糖正常,胰岛素、C肽、糖化血红蛋白均正常,不支持糖尿病诊断。(2)肾小管酸中毒:患儿婴儿期发病,存在生长迟缓、多尿、厌食等症状,血气分析示持续性代谢性酸中毒,血钾减低,X线示肾性佝偻病改变,临床考虑存在肾小管酸中毒。但患儿同时伴有糖尿、氨基酸尿、低钠血症、低镁血症等表现,不能单纯用肾小管酸中毒解释。(3)糖原累积病I型:糖原累积病I型是一种常染色体隐性遗传病。其中,糖原累积病Ia 型是由于G6PC基因突变使肝脏葡萄糖-6-磷酸酶缺乏所致。典型表现为婴幼儿期起病的肝大、生长发育落后、空腹低血糖、高脂血症、高尿酸血症和高乳酸血症等。糖原累积病Ib型是由于SLC37A4基因突变使葡萄糖-6-磷酸转移酶缺乏所致。患者除了有Ia型表现之外,还可有中性粒细胞功能障碍及数量减少的表现[13]。本例患儿存在生长发育迟缓、代谢性酸中毒需注意鉴别此症,但患儿生后无低血糖发作,无肝大表现,多次检测血糖均在正常范围,中性粒细胞无减少,不支持该诊断。(4)半乳糖血症:半乳糖血症是由于半乳糖代谢途径中酶的遗传性缺陷导致的一种代谢性疾病,高浓度的1-磷酸半乳糖和半乳糖醇在患儿晶状体及肝、肾、脑组织中存积导致患儿出现晶状体白内障及其他相关器官功能受损[14]。本例患儿存在生长迟缓、角膜大量结晶,需注意鉴别此病。但本例患儿进食乳类食品后无呕吐、腹泻等消化道症状,无智力发育落后,无黄疸、肝大、肝功能异常等表现,血、尿遗传代谢病筛查无半乳糖血症的相关阳性发现,不支持该诊断。(5)维生素D缺乏性佝偻病:本例患儿1岁6月龄,X线提示存在佝偻病骨改变,需注意鉴别维生素D缺乏性佝偻病。该病多见于婴幼儿,通常与维生素D 摄入不足及缺乏日照相关[15],主要临床表现为易惊、多汗、枕秃、骨骺变化及骨软化[16],通常不伴有肾脏及其他系统损害,本例患儿无日照不足、维生素D 摄入不足等相关诱因,血25 羟维生素D3检测正常,且除佝偻病表现外还伴有肾性范科尼综合征表现及眼部异常结晶,不能用维生素D缺乏性佝偻病解释。(6)酪氨酸血症I型:酪氨酸血症I型又称肝肾型酪氨酸血症,是一种与酪氨酸代谢障碍相关的遗传代谢病。病因与肝肾组织中酪氨酸代谢的终末酶延胡索酰乙酰乙酸水解酶缺陷相关。多数患儿于新生儿或婴儿期急性起病,肝脏受累明显;少数晚发型患者起病于1岁以后,主要表现为生长发育迟缓、进行性肝硬化、肾小管功能受损、低磷性佝偻病[17]。本例患儿存在生长发育迟缓、肾小管功能受损及佝偻病骨改变需与此症鉴别,但患儿肝功能正常,血、尿遗传代谢病筛查及基因检测结果均无酪氨酸血症的相关阳性发现,不支持诊断。(7)范科尼综合征:该病是由近端肾小管功能受损导致的多种中小分子物质重吸收障碍综合征,临床主要表现为近端肾小管酸中毒、肾性糖尿、氨基酸尿、磷酸盐尿(低磷血症)、碳酸盐尿和尿酸尿(低尿酸血症)、低钾血症和低钙血症等[18]。本例患儿存在生长发育迟缓、多饮多尿、糖尿、氨基酸尿、FEK 增高、TRP降低、低钾血症、低钠血症、低镁血症、代谢性酸中毒等表现,临床符合范科尼综合征特点。范科尼综合征的原因包含遗传及获得性两种:遗传性多见于某些先天性代谢性疾病,如胱氨酸贮积症、Wilson's 病、果糖不耐受等;获得性多见于暴露于某些毒素或药物等,以及多发性骨髓瘤、干燥综合征等继发因素。本例患儿除存在肾性范科尼综合征临床表现外,眼角膜可见大量胱氨酸结晶,基因检测CTNS基因c.922G>A点突变,考虑为遗传性胱氨酸贮积症所致。
4 诊断及确诊依据
诊断:胱氨酸贮积症(CTNS基因突变)。确诊依据:(1)临床表现:男性幼儿,婴儿期急性起病,表现为生长发育迟缓、尿糖、多饮多尿。(2)辅助检查提示患儿存在糖尿、氨基酸尿、FEK增高、TRP 降低、低钾血症、低钠血症、低镁血症、代谢性酸中毒,裂隙灯下观察到角膜结晶。(3)基因检测示CTNS基因存在可疑致病性纯合突变c.922G>A(p.Gly308Arg)。
5 讨论
胱氨酸贮积症是由于编码胱氨酸酶的CTNS基因变异,缺乏胱氨酸酶的溶酶体无法正常运转胱氨酸,胱氨酸蓄积并逐渐形成结晶,导致全身各系统受累。本例患儿为c.922G>A(p.Gly308Arg),目前尚无报道。
根据出现症状的年龄及肾脏受累的严重程度,胱氨酸贮积症分为3 种亚型:婴儿型、青少年型、眼病型。其中最为多见的是婴儿型,以肾脏受累为主要临床表现,通常表现为肾性范科尼综合征,肾小球功能进行性受损,最终会发展为肾功能衰竭。此型患者预后差,开始治疗时间较晚或未予治疗,通常在10 岁时就可进展为终末期肾病。青少年型,又称迟发型胱氨酸贮积症,患者病情较轻,进展较慢,通常不伴有明显的生长迟缓;多数患者病初仅表现为无症状蛋白尿及轻度肾性范科尼综合征,长期迁延后会出现明显的肾脏症状。眼病型患者通常无肾脏和其他器官症状,眼睛因胱氨酸晶体在角膜积聚,会出现畏光表现。本例患儿4月龄发病,急性起病,肾性范科尼综合征表现明显,考虑为婴儿型。
胱氨酸贮积症是一种系统性疾病,最常见、最严重的临床表现为肾损害;角膜上胱氨酸累积伴晶体形成是肾外首发症状;皮肤、内分泌和神经肌肉等其他系统均可出现相关的并发症[19-20]。由于胱氨酸在甲状腺、胰腺、性腺等分泌腺的异常蓄积可出现甲状腺功能低下、糖耐量异常、原发性性腺功能减退等临床表现;胱氨酸在神经系统蓄积可导致患者出现脑病、语言发育延迟、认知功能障碍、肌张力减低等神经系统症状[21-22]。目前可以通过检测白细胞中胱氨酸水平是否升高,对CTNS基因进行分子检测,通过裂隙灯下观察角膜胱氨酸晶体等方法对可疑患者进行确诊。本例患儿目前存在肾损害、角膜胱氨酸结晶,但甲状腺功能、血糖均在正常范围,智力发育正常,目前尚无皮肤、内分泌、神经肌肉等其他系统损害的相关依据。
对胱氨酸贮积症患者的治疗首先要注意维持机体内环境稳定,通过选择口服碳酸氢钠、柠檬酸钠钾溶液、枸橼酸钠钾溶液可以补充丢失的电解质、维持酸碱平衡;通过补给磷酸钠或磷酸钾及1,25 二羟骨化醇可以补充丢失的磷酸盐及维生素D3预防佝偻病发生;对甲状腺功能减低、生长迟缓、原发性睾丸功能衰竭、血浆睾酮浓度减低等内分泌疾病患者可以补充甲状腺素、生长激素、睾酮等相应缺乏激素进行替代治疗;对肾小球源性蛋白尿及肾小球滤过率下降患者可以应用血管紧张素转换酶抑制剂治疗;肾衰竭患者最终需进行肾移植治疗,对于肾功能持续不恢复患者待病情稳定后进行肾移植,抗肾抗体阳性患者需等待其转阴后再进行移植,移植前的终末期肾衰竭患者及肾移植后出现排异现象患者应行慢性透析。氨基硫醇半胱胺可以消耗体细胞和组织中溶酶体内蓄积的胱氨酸,是目前唯一针对胱氨酸贮积症的靶向治疗[23]。美国和欧洲分别于1994 年、1997年批准速释半胱胺酒石酸氢盐应用于胱氨酸贮积症的临床治疗[3],这是目前临床最常用的半胱胺,1 岁以内推荐治疗剂量1.30 g/(m·2d),能达到最佳疗效,且特异性清除胱氨酸治疗应维持终生。此外,目前造血干细胞移植及基因编辑技术也在积极开展,有望成为治疗胱氨酸贮积症的新方法[23]。在治疗初期,我们对患儿给予口服枸橼酸钠钾及肌内注射硫酸镁补充电解质、维持酸碱平衡,口服骨化三醇胶丸治疗佝偻病,同时口服补充左旋肉碱;确诊胱氨酸贮积症后,首次通过正规合法渠道由国外购入半胱胺酒石酸胶囊清除胱氨酸;目前我们正在监测患儿生长曲线,关注患儿生长发育落后情况,必要时可用生长激素治疗生长迟缓;此外,还将长期监测患儿电解质、血气分析等指标,监测肾功能、角膜及其他系统相关并发症情况。
综上所述,胱氨酸贮积症是一种罕见的遗传性疾病,容易误诊及漏诊。对于主要表现为范科尼综合征及眼部、神经、内分泌等多系统损害的患儿应注意鉴别。尽早应用半胱胺药物针对胱氨酸进行特异性清除,对延缓并发症出现,延长患者生命具有重要意义。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21 02:14:50
环境污染与防治(2021年11期)2021-01-09 02:08:51
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25 01:40:42
临床儿科杂志(2020年2期)2020-03-12 04:04:10
医学新知(2019年4期)2020-01-02 11:03:54
发明与创新(2019年42期)2019-11-18 01:24:04
农村青少年科学探究(2017年5期)2017-08-16 09:28:42
哈尔滨医药(2015年3期)2015-12-01 03:57:49
中国医疗美容(2015年4期)2015-04-27 02:24:11
现代检验医学杂志(2015年1期)2015-02-06 01:59:23
