
表面改性杂多酸衍生CoMoS催化剂的制备及其蒽加氢性能
2021-12-22 09:13陈彦飞陈卫娟孙广洵卢玉坤刘晨光柳云骐
石油学报(石油加工) 2021年6期
陈彦飞,陈卫娟,孙广洵,卢玉坤,刘晨光,柳云骐
(中国石油大学(华东)重质油国家重点实验室,山东 青岛 266580)
近年来,全球对于化石燃料,尤其是汽、柴油的需求有增无减,为此对于原油这种不可再生能源的高效利用提出了更高的要求。随着浅层优质石油资源的开发殆尽,劣质重油品质升级对贯彻“吃干榨净”的战略目标非常关键[1-4]。传统的重油加氢工艺,如固定床或沸腾床加氢工艺,对于原料品质要求较高,装置操作不够灵活,而浆态床加氢工艺对原料适应性强、装置一次性投资低,目前多采用非负载油溶性催化剂[5-6],避免了因载体孔道焦炭堆积导致的催化剂失活,延长了装置开工周期,节约企业的生产成本[7]。通过增加催化剂的分散性可提高金属活性相原子利用率,增加催化活性位点暴露。浆态床加氢裂化反应本质上是临氢热裂化反应,提高催化剂的加氢能力,可有效增加活性氢的数量,快速淬灭热裂化反应过程中的大分子碳氢自由基,减少自由基碰撞结焦,同时还可以抑制原料中胶质、沥青质的脱氢反应,增加轻质油收率[8-9]。因此,努力开发高分散、高加氢能力的低成本催化剂非常重要并且紧迫。
过去几十年时间里,钼基催化剂因其具有高活性、高抗毒能力、较低成本等优势被广泛应用于加氢脱硫[10-13]、加氢脱氮[14]、加氢精制[15]和电解水制氢[16]等领域。根据原子构型和层间堆积的不同,MoS2分为1T、2H和3R 3种晶体结构,其中2H-MoS2晶型最稳定,在加氢领域应用也最多[17]。研究发现[18-19],2H-MoS2中基面位由于吸附氢的能力太强,导致原子氢难以脱附,从而使基面位失去活化后续氢分子的能力,失去催化活性,所以,2H-MoS2中活性位主要集中在吸附氢能力较弱的边角位。因此,研究者们通过开发新的合成方法、控制合成条件等调节MoS2的形貌或结构,从而达到增加边角位活性位点暴露或增加硫空位[20]的目的。除了增加活性位点数量,添加金属助剂也可以从根本上提高MoS2的本征活性,CoMoS相和NiMoS相被证实具有比单金属Mo基催化剂更高的加氢活性[21-22]。简单的金属机械混合很难得到理想的协同效果,双金属杂多酸作为一种组成结构确定、双金属牢固结合的亚纳米尺寸无机金属盐,可以有效增强双金属的协同催化作用[23-24]。同时,采用合适的表面活性剂,容易对杂多酸表面进行改性[25],使其在有机溶剂中均匀分散。
笔者通过采用阳离子表面活性剂封装Co-Mo杂多酸,改变其表面极性,使其由水溶性变为油溶性,硫化后制备超分散、高协同的CoMoS催化剂,其合成示意图如图1所示。同时,采用蒽加氢反应作为模型反应,验证其加氢活性和长周期运行性能,以期为制备新型浆态床加氢催化剂提供指导和借鉴。
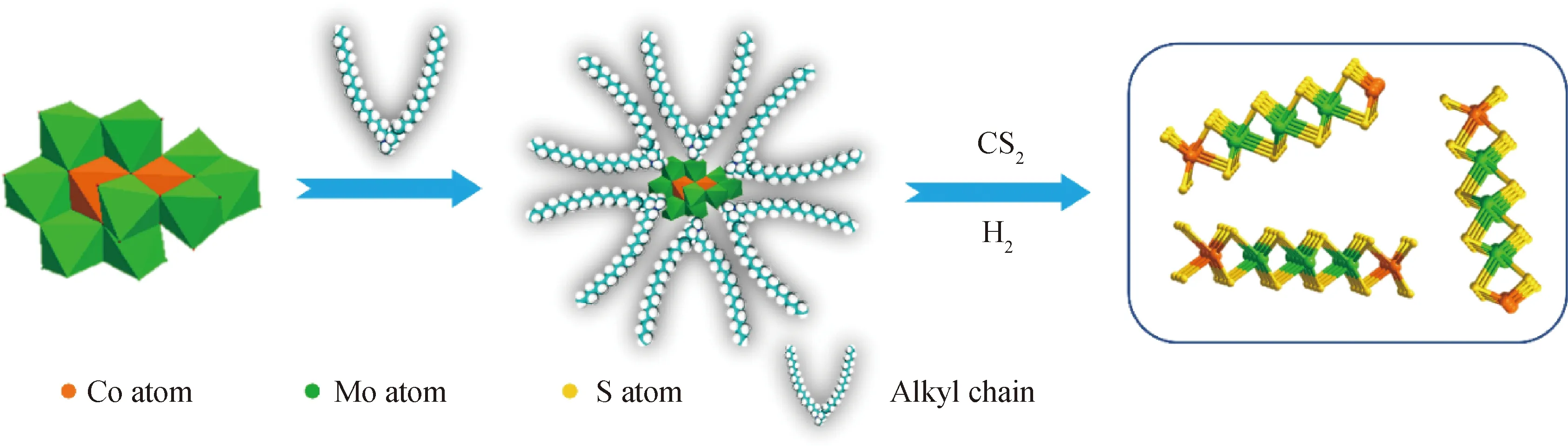
图1 CoMoS催化剂的合成示意图Fig.1 Schematic diagram of CoMoS catalyst synthesis
1 实验部分
1.1 原料与试剂
四水合钼酸铵((NH4)6Mo7O24·4H2O,AHM)、四水合乙酸钴(Co(CH3COO)2·4H2O)、活性炭、过氧化氢(H2O2),国药集团化学试剂有限公司产品;双十八烷基二甲基氯化铵([(C18H37)2(CH3)2N]Cl,DODAC)、三氯甲烷(CHCl3)、乙酰丙酮钼(C10H14MoO6,acMo)、乙酰丙酮钴(Ⅲ)(C15H21CoO6,acCo)、二硫化碳(CS2)、十三烷(C13H28)、蒽(C14H10,AN)、甲苯(C7H8),阿拉丁试剂公司产品;上述试剂均为分析纯,且未经过进一步纯化。去离子水为实验室自制。
1.2 Co2Mo10表面改性前驱体的合成
Evans-ShoweⅡ型杂多酸((NH4)6[Co2Mo10O38H4]·7H2O,Co2Mo10)按照文献中已报道的合成方法[26-27]进行合成。
随后使用阳离子表面活性剂DODAC对Co2Mo10进行表面改性。2.75 mmol DODAC溶于50 mL氯仿中,0.5 mmol Co2Mo10溶于50 mL去离子水中。搅拌条件下,氯仿溶液逐滴加入水溶液。搅拌1 h后,分液漏斗分离有机相,而后用50 mL去离子水洗有机相3次。利用40 ℃水浴或通风12 h除去氯仿,所得产物经40 ℃真空干燥处理获得Co2Mo10表面改性前驱体,并命名为Co2Mo10@DODA。
为了进行对比,选用乙酰丙酮钼(acMo)、乙酰丙酮钴(acCo)作为油溶性单金属有机钼和有机钴前驱体,同时将acMo和acCo按照5∶1的摩尔比机械混合作为油溶性双金属有机前驱体,并命名为acMo&acCo,分别参与后续硫化和反应。
1.3 催化剂的硫化
分别称取0.1 g前驱体、0.5 g CS2、40 g十三烷,加入250 mL不锈钢高压反应釜中进行催化剂的硫化反应。反应前使用H2置换釜内空气3次,当硫化温度为400 ℃、H2初始压力为7 MPa、搅拌速率为700 r/min时,反应1 h。反应后将反应釜快速空冷至室温。釜内产物经过滤、洗涤、60 ℃真空干燥,得到硫化态催化剂。为了防止硫化态催化剂因暴露于空气中被氧化,贮存在无水乙醇中,在表征前经过滤取出。Co2Mo10@DODA、Co2Mo10、acMo&acCo、acMo和acCo硫化后的催化剂分别命名为S-Co2Mo10@DODA、S-Co2Mo10、S-acMo&acCo、S-acMo和S-acCo。
1.4 催化剂的表征
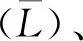
(1)
式中,Li代表片层i的长度,nm;xi代表具有片层长度Li的片层数量;
(2)
式中,Ni代表堆积层数;
(3)
式中,M(MoE)、M(MoC)和M(MoT)分别代表边位上、角位上和全部Mo原子数量,Mi代表MoS2边缘位上Mo原子的数量,由片层长度决定(Li=0.32(2Mi-1)nm)。
1.5 蒽加氢反应性能评价
为了充分发挥前驱体的油溶性,称取0.05 g Co2Mo10@DODA、0.5 g CS2、4 g蒽、40 g十三烷,加入250 mL不锈钢高压釜式反应器进行催化剂的蒽加氢反应性能评价;在反应温度为400 ℃、氢气初始压力为7 MPa、搅拌速率为700 r/min的条件下反应1 h。参比前驱体(Co2Mo10、acMo&acCo、acMo和acCo)按照与Co2Mo10@DODA相同的金属物质的量加入。进行了反应时间分别为1 min、2 h和3 h的蒽加氢反应,以采集实验数据和考察催化剂的长周期运行性能。
待反应釜降至室温,过滤得到反应后催化剂。采用装有氢离子火焰监测器的Agilent 7890 气相色谱仪(HP-5色谱柱)分析液相反应产物组成及比例。
蒽(AN)加氢产物主要包括二氢蒽(AH2)、四氢蒽(AH4)、八氢蒽(AH8)以及全氢蒽(AH14)。蒽加氢的反应路径图如图2所示[29]。采用蒽的转化率(x,%)、蒽的加氢率(h,%)[30]以及产物的选择性(s,%)评价催化剂的蒽加氢反应性能,分别按式(4)~(6)计算。
x=(ρ0-ρ)/ρ0×100%
(4)
s=AAHx/(AAH2+AAH4+AAH8+AAH14)×100%
(5)
h=x×(sAH2×2+sAH4×4+sAH8×8+
sAH14×14)/14×100%
(6)
式中,ρ0、ρ分别代表反应前、反应后蒽的质量浓度,g/mL;AAHx代表色谱谱峰面积;sAHx代表相应产物的选择性,%。
DHA—Dihydroanthracene;THA—Tetrahydroanthracene;OHA—Octahydroanthracene;PHA—Perhydroanthracene;OHP—Octahydrophenanthrene图2 蒽加氢的反应路径图Fig.2 Reaction path diagram of anthracene hydrogenation
2 结果与讨论
2.1 前驱体的结构和油溶性
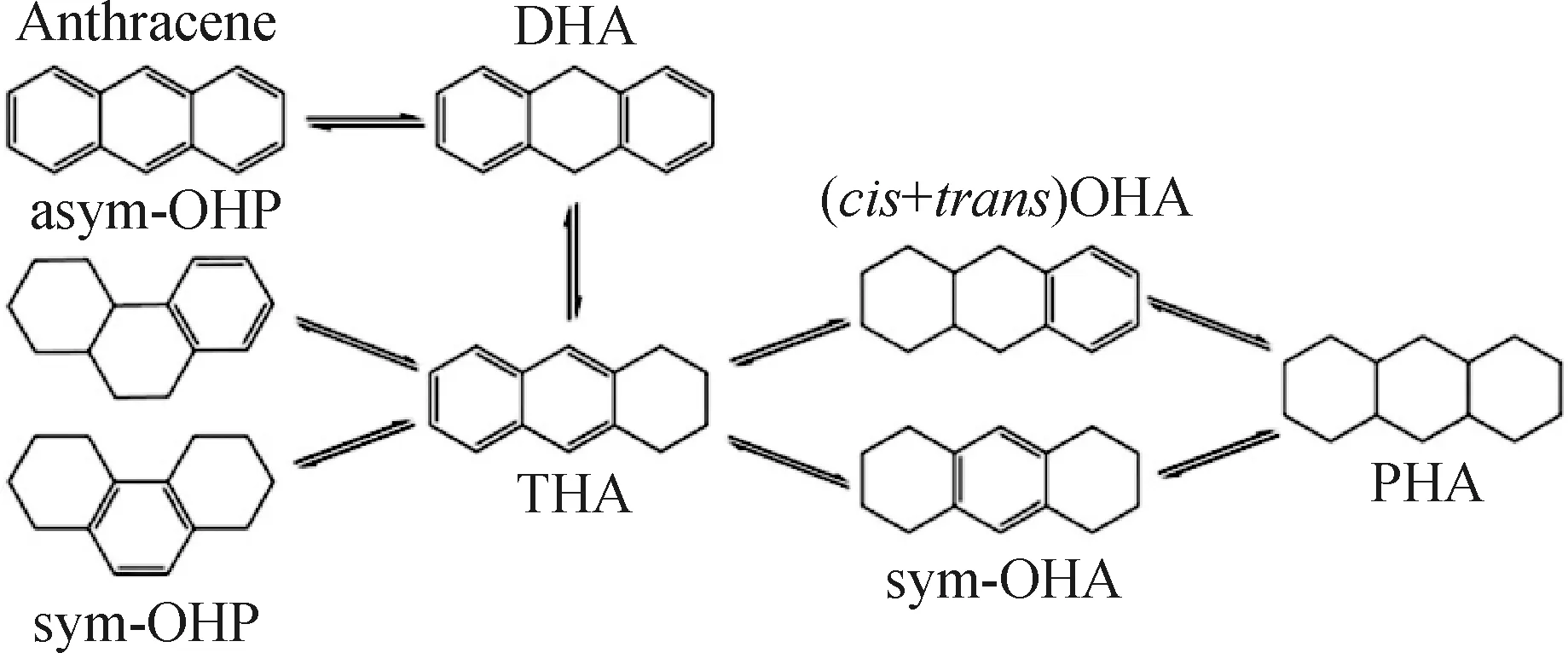
为了证实表面改性杂多酸的成功合成及其优秀的油溶性,采取一系列表征手段对其进行分析,结果如图3和表1所示。由图3中XRD谱图可以看到,Co2Mo10@DODA失去了Co2Mo10尖锐的晶体衍射峰,只表现出2个较弱的层状结构的堆积峰,表明DODA的引入破坏了Co2Mo10原有的晶体结构。从图3的FT-IR图中可以看到,Co2Mo10@DODA谱图中,3160、1403 cm-1处的N—H键振动峰消失,2956、2920、2851、1468、1378、721 cm-1处出现了C—H键的振动峰,表明NH4+被烷基链取代;同时,400~1000 cm-1波数范围内的Mo—O键的振动峰保持不变,说明Co2Mo10骨架结构没有被破坏。由图3中的TG图发现,Co2Mo10@DODA在700 ℃之前质量损失67.72%,与按照最终得到稳定氧化物的质量计算的理论质量损失量保持一致,由表1的Co2Mo10@DODA元素分析结果进一步确认了上述推测,从而得出Co2Mo10@DODA的化学式为[(C18H37)2(CH3)2N]6[Co2Mo10O38H4]·2H2O。
表1 Co2Mo10@DODA的元素分析结果Table 1 Elemental analysis results of Co2Mo10@DODA w/%
由图3中TEM照片可知,铜网上呈现均匀分布的黑点,采用ImageJ软件测量黑点粒径约为1 nm左右,与无机晶体学数据库中Co2Mo10阴离子的粒径一致,表明Co2Mo10@DODA经过表面改性,不仅具有极好的油溶性,同时可以实现在类沥青质溶剂甲苯中的单分子分散。上述表征结果表明,已成功合成表面改性杂多酸,更为重要的是实现了Co2Mo10@DODA在有机溶剂中的单分子分散,对于后续硫化、蒽加氢反应过程中减少MoS2片层聚集,增加活性位点暴露具有积极意义。
2.2 硫化态催化剂的组成和结构
在Co2Mo10@DODA硫化过程中,对升温到200 ℃的催化剂进行HAADF-STEM及XPS分析,结果如图4所示。从图4中可以发现,催化剂HAADF-STEM照片中呈现出Co2Mo10阴离子颗粒与MoS2单片层,同时照片中也呈现出两者混杂在一起的颗粒和单片层。图4(d)Mo 3 d的XPS谱图出现了Mo4+的信号峰,同时归属于Mo6+和Mo5+的XPS谱峰也没有完全消失,表明Co2Mo10@DODA在升温硫化过程中,Co2Mo10颗粒逐渐转变为MoS2片层结构。
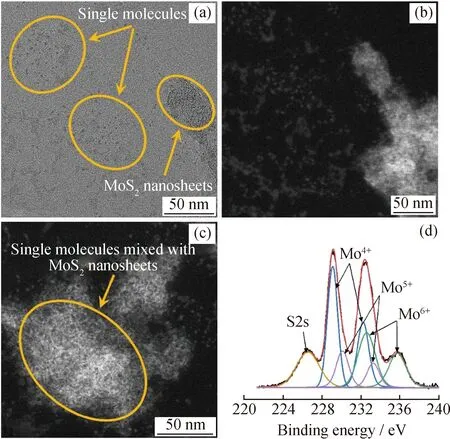
图4 Co2Mo10@DODA前驱体硫化过程的HAADF-STEM照片和元素Mo的XPS谱图Fig.4 HAADF-STEM images and Mo XPS spectra of the sulfurization process of Co2Mo10@DODA precursor(a)Brightfield image of isolated particles and nanosheets;(b)Darkfield image of isolated particles and nanosheets;(c)Darkfield image of mixed particles and nanosheets;(d)Mo 3 d XPS spectra (Co2Mo10@DODA samples were heated up to 200 ℃,and then it dropped to the room temperature)
图5为硫化态催化剂S-Co2Mo10@DODA及参比催化剂的XRD谱图。从图5中可以看出,3种催化剂均在2θ为33.1°和58.9°处出现了特征峰,分别归属于2H-MoS2的(110)和(100)晶面(JCPDS card No.37-1492)。其中,S-acMo&acCo在2θ为14.7°处出现了较明显的肩峰,归属于2H-MoS2的(002)晶面,表明S-acMo&acCo在c轴方向的片层堆叠层数可能较多。
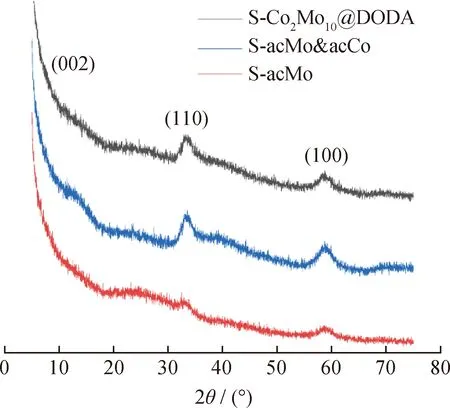
图5 S-Co2Mo10@DODA及参比催化剂S-acMo&acCo和S-acMo的XRD谱图Fig.5 XRD patterns of S-Co2Mo10@DODA and the reference catalysts S-acMo&acCo and S-acMo

图6 硫化态催化剂的TEM照片Fig.6 TEM images of sulfurized catalysts(a),(b)S-Co2Mo10@DODA;(c),(d)S-Co2Mo10;(e),(f)S-acMo&acCo;(g),(h)S-acMo;(i),(j)S-acCo;The insets of (a),(e),(g),(b),(f),and (h)are statistics results of the stacking layer number and the lengths of catalyst nanosheets,respectively;The inset of (j)is electron diffraction (SAED)image of the selected area

表2 TEM图中硫化态催化剂的MoS2片层数据统计表Table 2 Statistical table of MoS2 nanosheets of sulfurized catalysts in TEM images
图7是硫化态催化剂表面Mo元素的XPS谱图。由图7可知:根据谱峰曲线,Mo元素谱图可分为三组自旋轨道分裂峰,峰间距大约为3.2 eV[32]。Mo3 d5/2组分在(232.3±0.1)、(230.2±0.1)和(228.8±0.1)eV的谱峰分别归属于MoO3物种(Mo6+)、MoOxSy物种(Mo5+)和MoS2物种(Mo4+);在(226.02±0.1)eV的谱峰归属于硫物种(S2 s)。S-Co2Mo10@DODA、S-Co2Mo10、S-acMo&acCo 和S-acMo这4种催化剂表面Mo元素谱峰的分峰位置和数量相差不大。通过计算图7中谱峰面积得到不同价态Mo元素所占摩尔比,如表3所示。由表3可知,除了S-Co2Mo10中Mo的硫化度(Mo4+在所有价态中所占比例)明显较低(76.4%),其余硫化态催化剂中Mo6+、Mo5+比例基本相同,表明前驱体的油溶性对于Mo的硫化度有一定影响,进一步证实了TEM中所得结果。
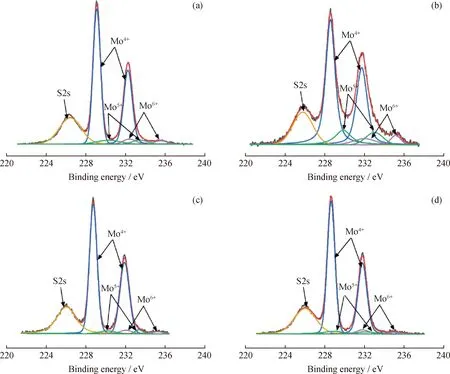
图7 硫化态催化剂元素Mo的XPS谱图Fig.7 XPS spectra of sulfurized catalyst element Mo(a)S-Co2Mo10@DODA;(b)S-Co2Mo10;(c)S-acMo&acCo;(d)S-acMo
图8是硫化态催化剂表面Co元素的XPS谱图。对硫化态催化剂Co元素的XPS谱图进行分峰拟合处理,也可得到三组自旋轨道分裂峰和一些震激峰。由图8可知:在结合能(797.5±0.1)和(781.5±0.1)eV处呈现出CoOx的2p1/2和2p3/2的谱峰,在结合能(793.2±0.1)和(778.2±0.1)eV处呈现出CoSx的2p1/2和2p3/2的谱峰,在结合能(778.8±0.1)和(793.9±0.1)eV处呈现出CoMoS的2p1/2和2p3/2的谱峰。此外,4种催化剂CO的XPS谱图还包含4个震激峰。对于双金属Co-Mo催化剂来说,起到主要加氢活性的催化位点位于边角位的CoMoS相。由表3可知,硫化态催化剂中,S-Co2Mo10@DODA中CoMoS所占摩尔分数最高为56.9%,S-Co2Mo10中CoMoS所占摩尔分数最小为31.4%,说明S-Co2Mo10@DODA有可能具有最高的加氢催化活性。在S-acCo(图8(d))中,未发现CoMoS相,但是存在大量的CoSx(摩尔分数为43.9%),与TEM中的结果一致。
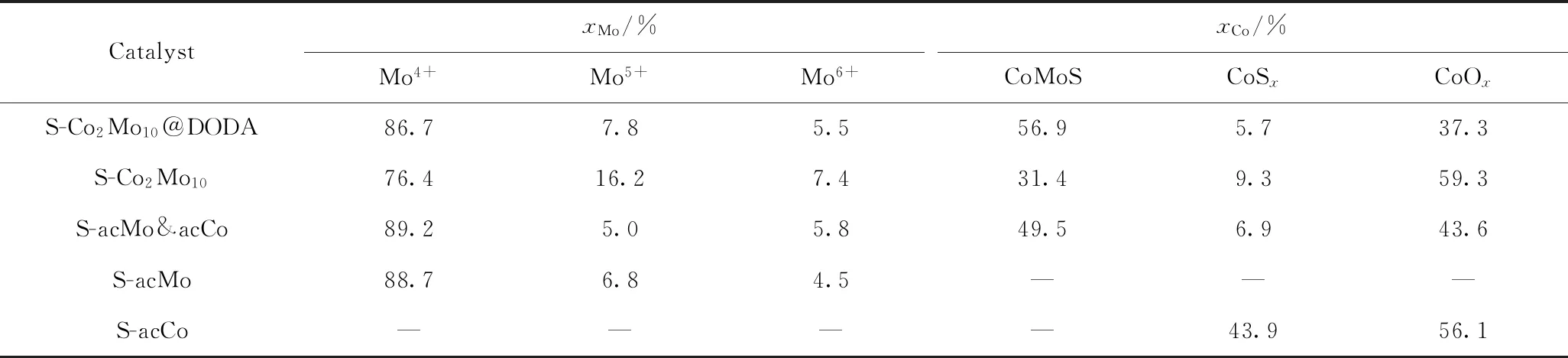
表3 硫化态催化剂的XPS分析数据表Table 3 XPS analysis data sheet of sulfurized catalysts
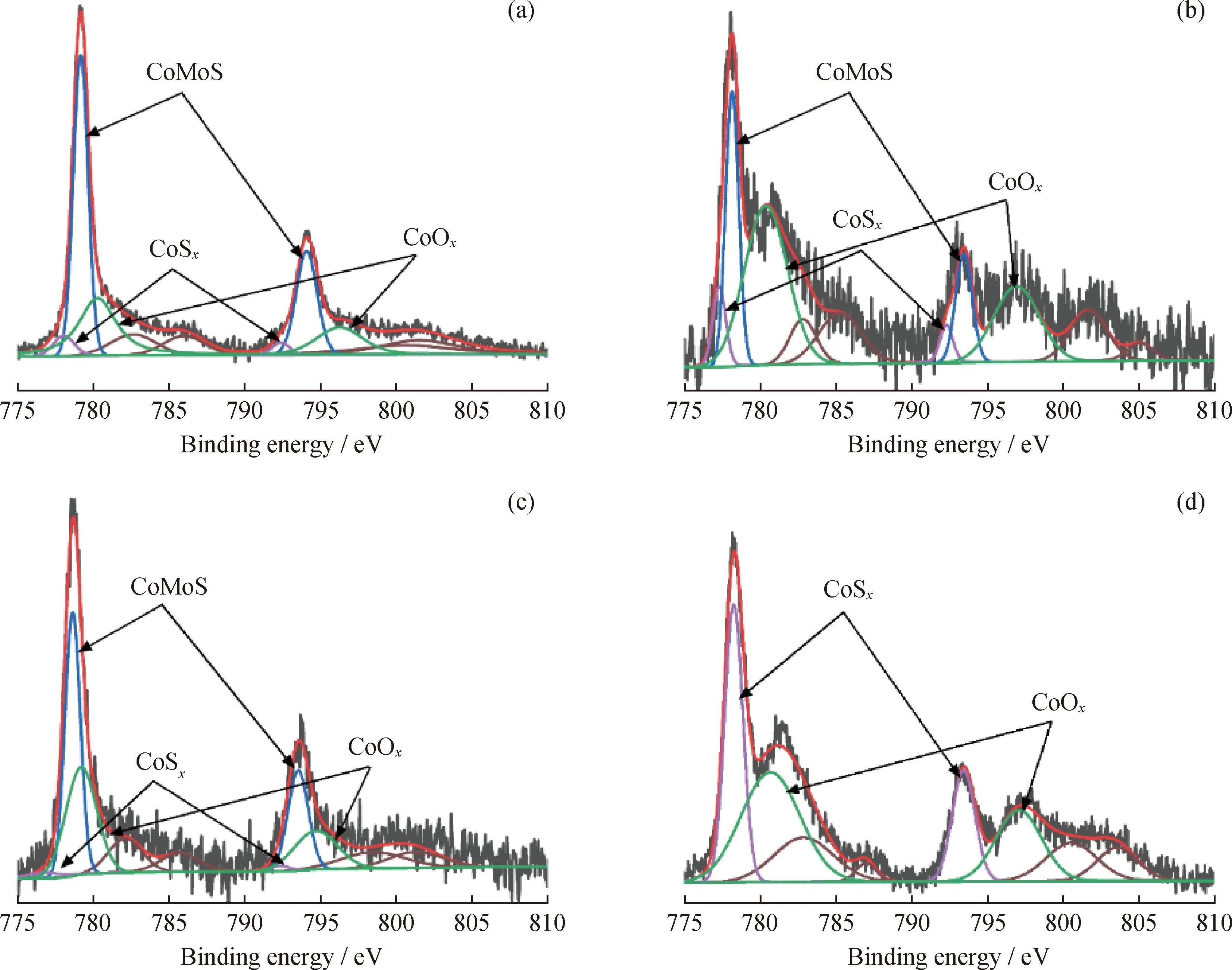
图8 硫化态催化剂元素Co的XPS谱图Fig.8 XPS spectra of the sulfurized catalyst element Co(a)S-Co2Mo10@DODA;(b)S-Co2Mo10;(c)S-acMo&acCo;(d)S-acCo
综上所述,在经过表面改性后,前驱体Co2Mo10@DODA由亲水性转变为亲油性,同时在甲苯等有机溶剂中可以实现单分子分散(图3(d)),从而可将杂多酸成功引入到有机体系中,拓宽了杂多酸这类无机酸盐的应用范围。正是由于Co2Mo10@DODA可以在有机溶剂中实现单分子分散,使得其与有机钼/钴混合物相比,分散性更好,硫化后MoS2片层具有更小的尺寸、更少的堆积层数、更好的分散性以及更高的硫化度,同时可以在一定程度上减少团聚,从而暴露更多的边角活性位,提供更多的活性位点,促使加氢反应加速进行。因此,表面改性的催化剂前驱体分散性可以有效影响硫化态活性相的聚集程度,杂多酸外包裹的一层烷基链在这个过程中也可能起到一定的积极作用(限域作用)。与此同时,与S-acMo&acCo相比,S-Co2Mo10@DODA具有更多的CoMoS活性相,这是因为Co2Mo10杂多酸阴离子中CoMo之间的化学键合,更有利于硫化过程中CoMo之间的紧密作用,从而形成更多的活性相,这远不是CoMo机械混合能相比的。极佳的油溶性和双金属Co-Mo之间的紧密作用使得Co2Mo10@DODA在硫化后生成了催化活性相尺寸小、分散性好的CoMoS催化剂。
2.3 催化剂的蒽加氢性能
对于浆态床加氢裂化反应来说,催化剂可以有效抑制减压渣油(VR)中不饱和组分加氢反应中的结焦,提高轻质油收率,因此,提高加氢裂化催化剂的加氢能力具有实际应用价值。笔者采用模型化合物蒽(AN),考察硫化态催化剂S-Co2Mo10@DODA及参比催化剂对于稠环芳烃的加氢效果。蒽加氢反应是一个逐步加氢的过程(如图2所示),其加氢难度随着加氢反应的深入快速增加。加氢反应温度较高时,甚至会发生全氢蒽的脱氢反应,因此对于催化剂的加氢能力提出了很高的要求。图9是催化剂的蒽加氢评价性能图。由图9可知:当不加催化剂反应时,蒽在400 ℃的转化率已经达到98.8%,但是主要产物为二氢蒽和四氢蒽,八氢蒽的选择性只有28.5%,没有全氢蒽生成。使用硫化态催化剂S-Co2Mo10@DODA 和S-acMo时,蒽的转化率均达到了100%。值得注意的是,使用硫化态催化剂S-Co2Mo10和S-acCo时蒽的转化率均低于不加催化剂时,推测原因是,使用这2种催化剂的反应过程中生成的CoSx物种对蒽产生了吸附作用,两者之间发生电子转移,提高了蒽加氢反应发生的能垒,从而降低了加氢的反应速率。在选择性上,同样是使用硫化态催化剂S-Co2Mo10@DODA和S-acMo时的深度加氢产物质量分数更高,尤其是S-Co2Mo10@DODA 的八氢蒽选择性达到82.3%,全氢蒽选择性也达到2%,加氢效果最好。基本上,使用催化剂时蒽的转化率高,对应反应的深度加氢产物选择性就大。为了能更清晰地比较催化剂的加氢性能,笔者引入加氢率[28]的概念(图9(a)),加氢率(h,%)代表的是产物消耗分子氢的多少。使用硫化态催化剂S-Co2Mo10@DODA的蒽加氢率达到55.5%,远远超过其余参比催化剂。可见,与油溶性钼钴前驱体和未改性杂多酸相比,Co2Mo10@DODA作为前驱体所生成的硫化态催化剂具有非常高的蒽加氢活性。
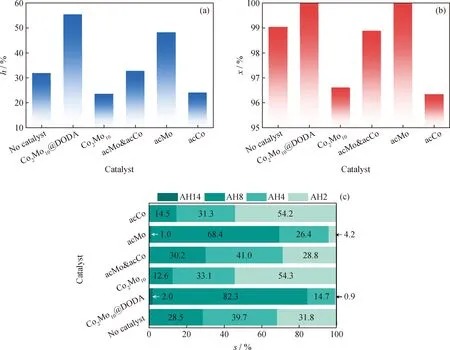
图9 催化剂的蒽加氢评价性能图Fig.9 Performance evaluation charts for anthracene hydrogenation of catalysts(a)Hydrogenation percentage (h);(b)Conversion rate (x);(c)Selectivity (s)Conditions:T=400 ℃;p=7 MPa;t=1 h
对于大多数催化剂来说,提高其催化活性的方式有2种:一种是增加催化活性位数量,一种是提高单位活性位本征活性。硫化态催化剂S-Co2Mo10@DODA加氢催化活性高正是来源于其具有的这两种特性:(1)MoS2片层尺寸小,堆积层数少,分散度高,带来的优势是催化剂会暴露更多的边角活性位;(2)S-Co2Mo10@DODA 催化剂本身具有更高比例的CoMoS相。这两种特性相互促进,使得催化剂的加氢活性得以大幅度提高。表面活性剂改性策略增加了杂多酸与有机溶剂的相容性,使其可以单分子分散在有机溶剂中,极大的减小了前驱体分散单体的尺寸,受此影响,硫化后催化剂颗粒的尺寸也明显减小,边角活性位数量显著增多。同时,在硫化过程中,表面活性剂可以发挥一定的限制MoS2片层增长以及抑制其聚集的作用。另一方面,引入了杂多酸Co2Mo10的Co2Mo10@DODA前驱体,在Co2Mo10的阴离子中,金属Mo和Co以化学键链接,原子之间距离近,相互作用强,这样在硫化后就会生成更大比例的CoMoS相,避免生成本征催化活性较低的单金属硫化物。因此,笔者的催化剂设计策略对于进一步提高Mo基硫化物催化剂的加氢活性具有积极的意义。
为了研究长周期反应过程中蒽加氢产物的变化规律,使用硫化态催化剂S-Co2Mo10@DODA进行3 h的蒽加氢反应。图10为蒽加氢反应中反应釜内压力随时间的变化曲线图;图11为蒽加氢反应中,转化率、加氢率和产物选择性随反应时间变化的曲线图。由图10可知,在反应升温过程中,釜内压力由7 MPa急速升到13 MPa左右,并在反应过程中基本维持稳定。由图11(a)可知,在反应釜的升温过程中,蒽的加氢转化率和产物加氢率均快速增长。实际温度到达设定温度400 ℃后,蒽的加氢转化率在达到99.6%后首先出现拐点,进入缓慢增长期,并在反应进行到1 h时就达到100%的加氢转化率。而加氢率的拐点出现在反应1 h的时候,后续随着反应时间延长,加氢率缓慢增加,表明深度加氢产物八氢蒽和全氢蒽逐渐生成。由图11(b)可知,在产物选择性上,反应温度达到350 ℃时,二氢蒽的选择性非常高(92.6%),随后快速下降,在反应进行到1 h后接近消失,表明蒽加氢首先生成二氢蒽,随着反应温度升高迅速被转化为深度加氢产物。同时,四氢蒽作为反应中间产物,在选择性上也是先增加后减少,1 h后维持在一个较低水平(11%左右)。八氢蒽的选择性初始增加速率比四氢蒽稍慢,在其选择性达到最大值后,出现缓慢下降。而最终产物全氢蒽在反应升温阶段基本不生成,在温度达到400 ℃之后才出现,并随着反应时间延长缓慢而坚定的增加。可见,蒽加氢反应是一个逐步加氢的过程,与文献中的描述一致[29],同时深度加氢产物中的全氢蒽在400 ℃时选择性较低。
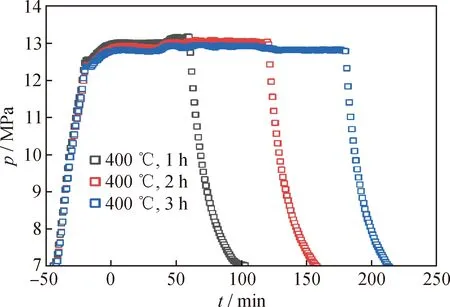
图10 催化剂S-Co2Mo10@DODA参与的蒽加氢反应中压力随时间的变化曲线图Fig.10 Pressure change curves of anthracene hydrogenation reactions over time with the participation of S-Co2Mo10@DODA catalyst
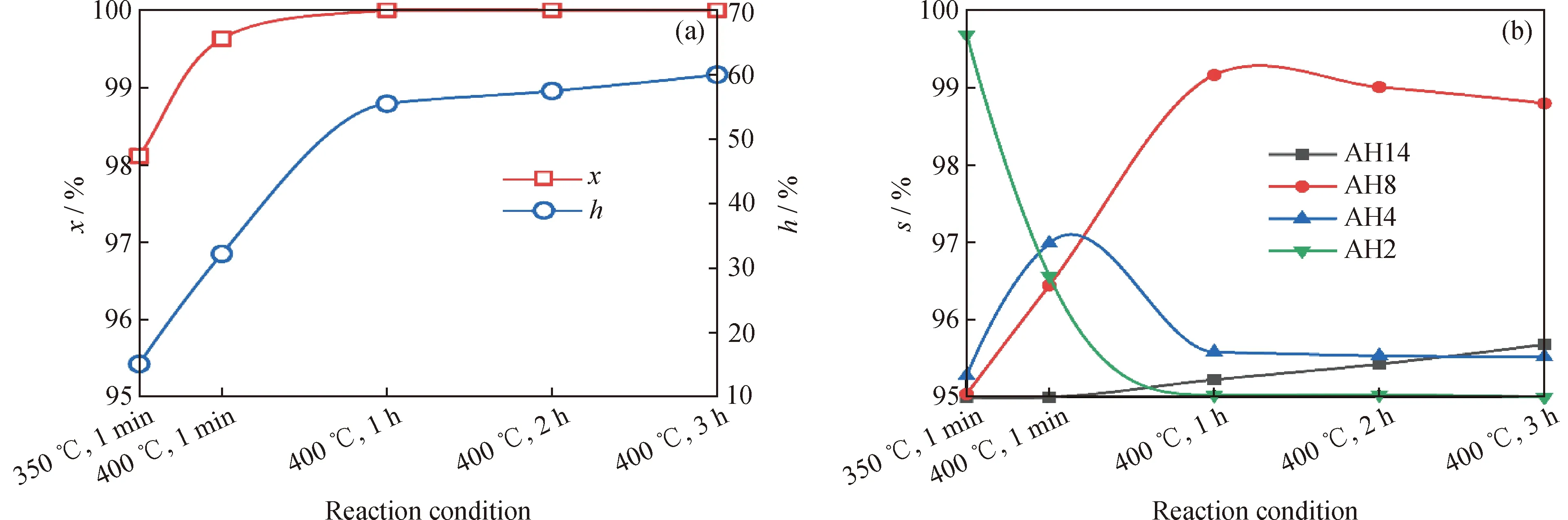
图11 催化剂S-Co2Mo10@DODA参与的蒽加氢反应中性能曲线图Fig.11 Performance chart for anthracene hydrogenation reactions with the participation of S-Co2Mo10@DODA catalyst(a)Conversion rate (x)and hydrogenation percentage (h);(b)Selectivity (s)Condition:p=7 MPa
2.4 催化剂的长周期运行性能
图12是不同反应时间后硫化态催化剂S-Co2Mo10@DODA 的XRD谱图。由图12可知,在反应时间逐渐延长的过程中,硫化态催化剂S-Co2Mo10@DODA 中的MoS2微晶衍射峰并没有发生明显变化,以2H-MoS2的(110)晶面和(100)晶面衍射峰为主,同时还有微弱的(002)晶面衍射峰。在反应1 min后的样品在2θ为26.4°处出现一个小尖峰,归属于无定形碳的衍射峰,推测在反应初期,硫化态催化剂S-Co2Mo10@DODA中的烷基碳链尚未完全脱落,仍然吸附在催化剂上。
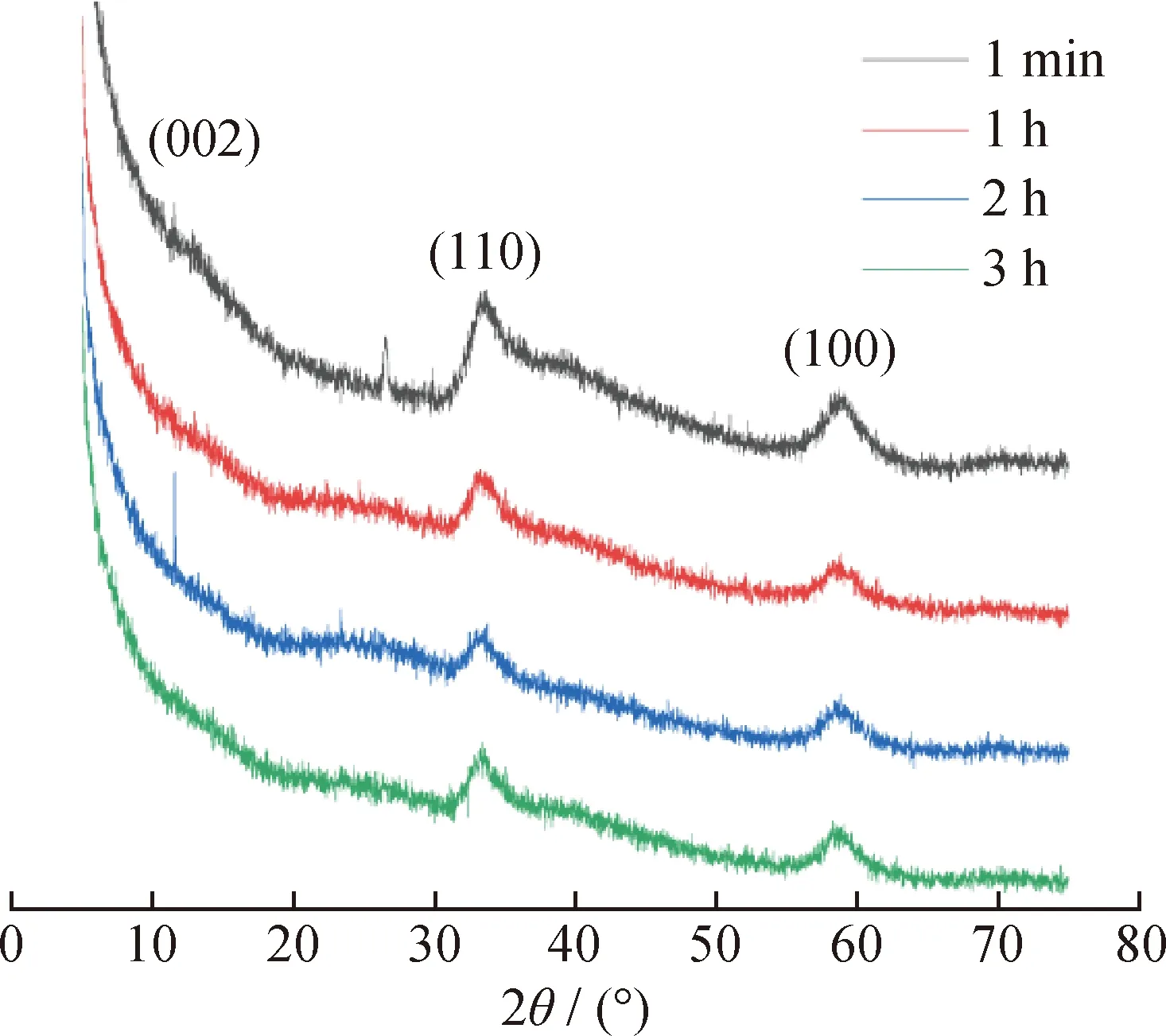
图12 不同反应时间后催化剂S-Co2Mo10@DODA的XRD谱图Fig.12 XRD patterns of S-Co2Mo10@DODA catalyst after different reaction time

图13 不同反应时间使用后的催化剂S-Co2Mo10@DODA的TEM图Fig.13 TEM images of catalyst S-Co2Mo10@DODA after different reaction time(a),(b)1 min;(c),(d)1 h;(e),(f)2 h;(g),(h)3 h;The insets of (a),(c),(e),(g)are statistics results of the stacking layer number;The insets of (b),(d),(f),(h)are statistics results of the lengths of catalyst nanosheets.
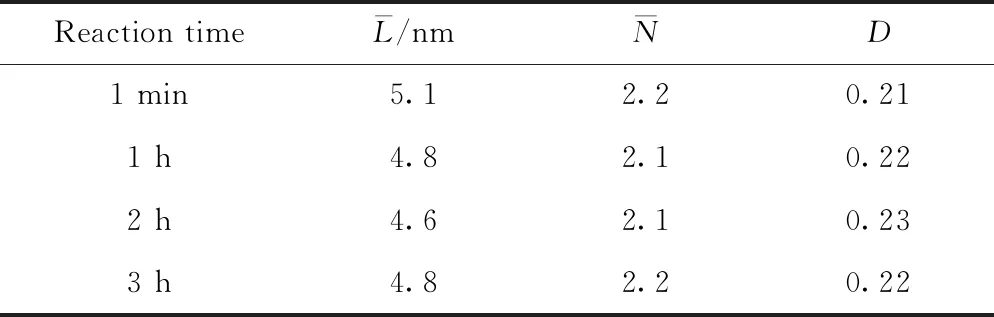
表4 不同反应时间后TEM照片中催化剂S-Co2Mo10@DODA的MoS2片层数据统计表Table 4 Statistical table of MoS2 nanosheets of S-Co2Mo10@DODA catalyst after different reaction time as shown in TEM images
对蒽加氢不同反应时间后S-Co2Mo10@DODA催化剂表面的金属元素的价态分布和摩尔比采用XPS进行分析,分峰处理后S-Co2Mo10@DODA中不同价态元素Mo、Co的摩尔比数据列于表5。由表5可知,Mo、Co元素价态的分布范围和比例并没有随蒽加氢反应时间的延长发生变化,且相关数据与表3中S-Co2Mo10@DODA的数据差别在误差允许范围之内。
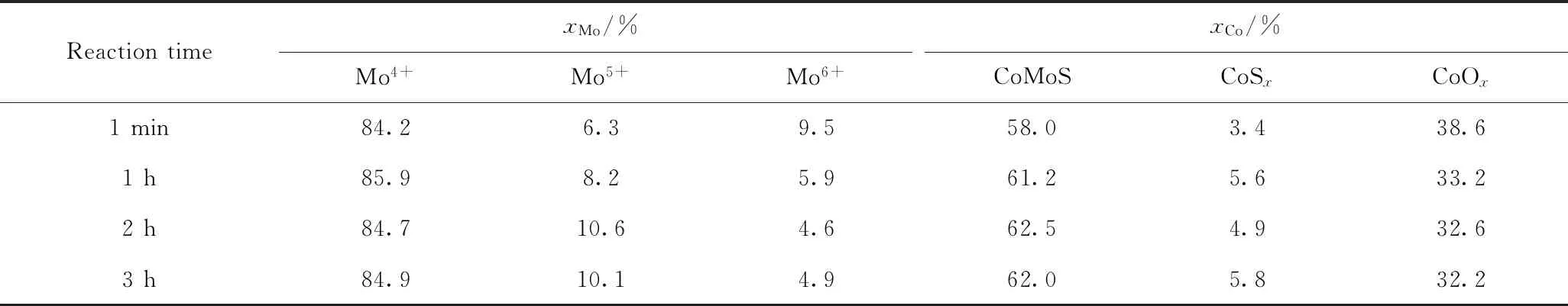
表5 不同反应时间使用后催化剂S-Co2Mo10@DODA的XPS分析数据表Table 5 Statistical table of XPS spectra of S-Co2Mo10@DODA catalyst after different reaction time
综上所述,在蒽加氢反应过程中,硫化态催化剂S-Co2Mo10@DODA活性相的形貌和组成并没有随反应时间的延长发生变化,表明硫化态催化剂S-Co2Mo10@DODA在蒽加氢反应中具有良好的长周期运行性能。
3 结 论
(1)引入Co2Mo10杂多酸,并采用阳离子表面活性剂DODAC对杂多酸表面进行改性,利用静电相互作用力成功实现对Co2Mo10的封装。合成的前驱体Co2Mo10@DODA具有良好的油溶性,能以单分子的形式分散在有机溶剂中,在较低温度下(200 ℃)即可被硫化。
(2)由于Co2Mo10@DODA前驱体的超分散特性和Co2Mo10中Co、Mo之间的键合作用,硫化态催化剂S-Co2Mo10@DODA中的MoS2片层具有更小的尺寸、更低的堆叠层数和更好的分散性,MoS2片层可暴露更多的边角催化活性位,同时具有更高比例的CoMoS相。
(3)硫化态催化剂S-Co2Mo10@DODA在蒽加氢反应中具有优秀的催化活性,在400 ℃、氢初压为7 MPa的条件下反应1 h,蒽转化率达到100%,深度加氢产物(八氢蒽和全氢蒽)选择性达到84.3%。
猜你喜欢
弹性体(2022年3期)2022-11-15
科学与财富(2021年33期)2021-05-10
陶瓷学报(2020年2期)2020-10-27
陶瓷学报(2020年2期)2020-10-27
矿产综合利用(2020年1期)2020-07-24
天津医科大学学报(2019年3期)2019-08-13
中国特种设备安全(2019年3期)2019-04-22
汽车零部件(2018年5期)2018-06-13
中国资源综合利用(2017年4期)2018-01-22
山东工业技术(2016年15期)2016-12-01
