
先天性肺气道畸形患儿病变组织中差异表达基因的筛选及生物学功能分析
2021-12-18 11:35张刚刘丹丹蔡纯李潇曾慧仪娄蕾闫宪刚俞钢
山东医药 2021年33期
张刚,刘丹丹,蔡纯,李潇,曾慧仪,娄蕾,闫宪刚,俞钢
1广州医科大学附属第三医院小儿外科,广州510150;2广州医科大学附属第三医院超声医学科
先天性肺气道畸形(CPAM)在先天性呼吸系统畸形中最常见,占比30%~40%,患病率占出生活胎的1/35 000~1/7 200[1],近年来呈明显上升态势,这与CPAM产前诊断技术的进步有一定关系[2]。当前,多数患儿可在孕18~22周时通过产前超声筛查被发现,敏感度达94%,特异度达95.3%[2]。CPAM的发病机制目前尚不明确,可能是由于胚胎发育过程中肺部上皮细胞与下层间充质细胞之间的信号传导障碍,导致病变中缺乏正常的肺泡,进而形成以终末细支气管过度增生与扩张为特征的错构瘤样病变。研究[2-4]显示,肺胚胎发育涉及多种生长因子、转录因子和信号分子在上皮—间充质的相互作用,以及信号调控分子在肺上皮细胞和间充质细胞之间的相互作用。当前,二代测序技术所得数据稳定可靠,价格相对低廉,已广泛应用于基础研究、临床诊断和药物研发等领域。此技术可快速获取某一物种特定器官或组织在某一状态下几乎所有转录本的序列信息,进行疾病与正常样本间的基因表达差异分析、可变剪切和融合基因分析,进而寻找致病基因、探索发病机制。相对于传统基因芯片而言,二代测序技术无需预先设计特异性探针,具有检测范围广、准确率及分辨率高等优点[5]。本研究利用二代测序技术结合生物信息学分析,筛选了CPAM患儿病变组织中的差异表达基因,并对差异表达基因进行生物信息学分析,为深入研究CPAM的发病机制提供新思路。
1 资料与方法
1.1 临床资料 选取广州医科大学附属第三医院小儿外科2020年1—3月收治的CPAM患儿5例。纳入标准:①产前检查、术前CT检查、术中诊断、术后病理检查均确定为1型CPAM;②患儿年龄0.5~1岁,病灶直径5~10 cm,无胎儿水肿、术前无感染、未接受化疗和放射治疗、无合并明确的染色体畸形等情况;③患儿接受胸腔镜微创手术治疗,术后未留置胸管,无感染、出血、再手术等,随诊1年未见存留、复发、再发、继发感染、气胸等;④患儿监护人均签署知情同意书并支持本研究。5例患儿术中分别留取CPAM病变组织、CPAM病变旁组织(直径5 mm)各1份,共计10个样本,分别标记为1a、1b、2a、2b、3a、3b、4a、4b、5a、5b,样本置于液氮中冷冻后迅速转移至-80℃冰箱中保存。
1.2 CPAM患儿病变组织中差异表达基因的筛选
1.2.1 受检样本RNA-Seq文库的构建 采用TRIzol法对样本总RNA进行抽提,抽提所得的总RNA取样分别进行浓度、28S/18S或23S/16S、RIN值质检,质检合格后,使用Total RNA-seq(H/M/R)Library Prep Kit for Illumina®构建文库:先将设计好的DNA探针与RNA样品进行杂交,从而将rRNA从总RNA中去除;随后将RNA进行片段化,合成cDNA第一链,采用链特异的方法,在第二链cDNA合成时掺入dUTP对其进行标记,同时在此步完成末端修复;接着进行加A-尾、连接接头、连接产物纯化和片段大小分选、文库扩增等步骤,在PCR扩增前用UDG酶消化带dUTP的第二链模板,PCR扩增结束后用磁珠纯化回收,即得RNA-Seq文库。
1.2.2 RNA-Seq文库质量评估 文库构建完成后,先使用Qubit 3.0进行初步定量,随后使用Agilent 2100 Bioanalyzer对文库进行检测,插入的目的片段大小符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>3 nM),以保证文库质量[5]。
1.2.3 RNA-Seq文库的测序 文库质量评估合格后,将不同文库按照有效浓度及目标下机数据量的需求测序(测序平台:Novaseq 6000 PE150,美国Illumina®公司),测序数据量为10 G:对原始序列进行过滤,去除接头序列、含N较多的序列及低质量的序列,获得待分析序列,用Fast QC软件对其进行质量评估[6]。通过与核糖体数据库进行比对,去除核糖体RNA序列,用RSEM软件[7]获得基因的表达序列数。只有表达丰度足够高的基因才是具有生物学意义的差异表达基因,反应真实的生物学现象,因此,需要对基因的表达进行了过滤。过滤标准为:基因的表达值(CPM值)至少在一半样本中都大于1才被保留。然后,使用M-值的加权截尾均值法(TMM)对不同样本每个基因的表达进行标准化,获得有效序列。
1.2.4 异常样本剔除 利用主成分分析来考察样品中是否存在异常样品:通常组内的样本会聚类在一起,组间的样本会分隔开,异常样本则会和本组内样本分隔开。选择两个主成分因子:分别从序列差异性最大PC1(63.6%)和次大PC2(13.4%)两个方向提取,制作主成分分析图。如果检测到异常样本,在差异分析时,该样本应该被排除在外。
1.2.5 CPAM病变组织与CPAM病变旁组织差异表达基因的提取与筛选 剔除异常表达的样本后,使用edgeR软件将有效序列与参考基因组进行比对、序列预测、表达值计算,设置差异倍数foldchange=1.5、P<0.05、FDR<1,筛选出CPAM病变组织与CPAM病变旁组织的差异表达基因[8]。1.3 差异表达基因的基因本体(GO)功能富集分析和KEGG信号通路分析 使用R软件cluster Profiler对差异表达基因进行分析[9]:通过计算差异表达基因与GO集合的交集获得统计学显著性(超几何检验,P=0.05)来判断目标基因集合是否能发挥某种功能,包括差异表达基因参与的生物学过程、细胞组成以及分子功能三个层面,取P<0.05且P值最显著的前10个结果。利用cluster Profiler在线网站对筛选的差异基因注释到最新的KEGG信号通路进行分析,按照P<0.05筛选出排名前10的信号通路。
2 结果
成功构建10个样本的RNA-Seq文库,文库质量合格,各样本序列数见表1。
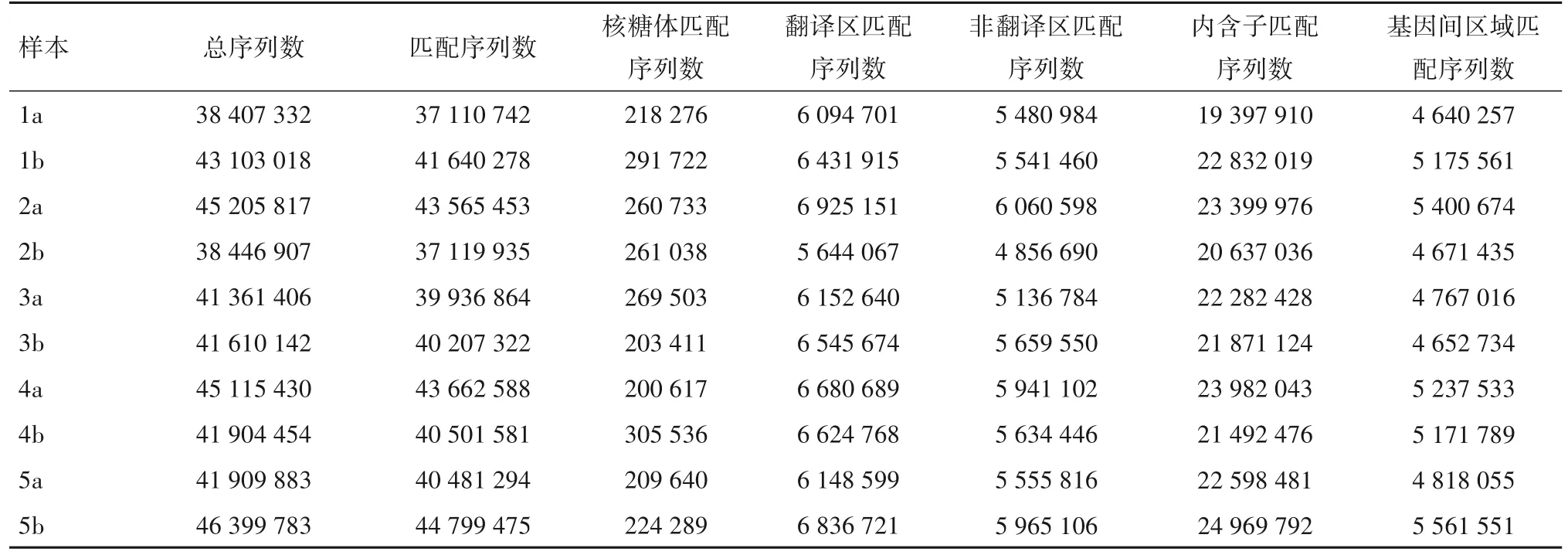
表1 各样本序列数(个)
2.1 CPAM病变组织与CPAM病变旁组织差异表达基因的筛选结果 依据主成分分析结果,剔除异常表达的样本3b后,共筛选出CPAM病变组织与CPAM病变旁组织差异表达基因1 063个,其中上调的基因860个、下调的基因203个。上调的基因包括FOXJ1、Sox2、BMP8A、BMPR1B、DNAAF4、LRP2、MNS1、WT1、CRISP3、SCGB1A1、FCGBP、MSMB、SLC5A5、SPINK1、KRT17、CLCA2、UPK1B、UGT1A6、VTCN1、B3GNT3、ERN2、CCNO、KRT15、CXCL13、SPATS1、KRT5、MUC4、RHOV、RPL13AP17、MUC16、CYP2F1、SRD5A2、TMEM130、KLK13、PLEKHS1、EPN3、CDC20B、C1orf141、CCL11、RUNDC3B、SPP1、SPATA4、FUT2、TCP11X2、ANKRD66、CRISP2、CP、C12orf74、PROM1、PRR18等,下调的基因包括BMP6、BMPR2、BMPER、HBA1、Smad6、Smad7、LPL、CD52、TNFSF10、CD109、NAGS、NRG3、RAMP2、GPER1、FPR2、GBP1、C1QTNF5、WNT2B、CORO6、MFAP3L、BMPR2、EHD3、RSAD2、CRMP1、LILRA1、BMPER、MASP1、SEMA6A、DSC2、SLC16A6、ACSS3、ADAMTS7、CHRD、ZNF331、WNT2、EXOC3L1、GRIA1、ACVRL1、SMAD7、AATBC、TAL1、LOC400685、CCDC68、ABCA3、COL4A2、B3GNT8、MS4A14、DPEP2、LIN7A、GBP4、ITGA1等。
2.2 差异表达基因的GO功能富集分析和KEGG信号通路分析结果 GO功能富集分析结果显示,差异表达基因的细胞组成主要位于轴丝、纤毛质、活动纤毛、微管、微管相关复合物、细胞顶端部分、含胶原的细胞外基质、神经元细胞体、纤毛基体等,参与的分子功能主要有微管蛋白结合功能、受体配体活性、信号受体激活剂活性、内肽酶活性、ATP酶活性、肌动活性、微管结合功能、G蛋白耦联受体结合功能、微管运动活性、丝氨酸型肽酶活性等,涉及的生物学过程主要有纤毛组织、纤毛组装、基于微管的运动、纤毛运动、微管束形成、多细胞机体稳态、纤毛或鞭毛依赖性细胞运动、纤毛依赖性细胞运动、轴丝组装、鞭毛运动等。KEGG信号通路分析结果显示,差异表达基因参与了PI3K-Akt信号通路、血管平滑肌收缩、流体剪切应力和动脉粥样硬化、神经活性配体—受体相互作用、细胞因子—受体相互作用、亨廷顿病、转录失调、黏着斑、ECM-受体相互作用、病毒蛋白与细胞因子—受体相互作用、蛋白质消化吸收等信号通路的调控。
3 讨论
CPAM根据病理表现分为5型:0型表现为先天性肺泡发育不良,实性外观,肺缩小变硬,占1%~3%;1型由1个以上囊肿组成,壁厚,囊肿直径2~10 cm,占60%~70%;2型多个小囊组成,囊肿直径0.5~2 cm,占10%~15%;3型由蜂窝状微囊组成,囊肿直径<0.5 cm,约占5%;4型多为单个或分隔大囊,囊肿直径>10 cm,约占28%。本研究为保证样本量充足、研究结果的普惠性,选取发病率最高的1型CPAM样本为研究对象。
CPAM的病因尚无统一认识,当前主要有2种假说[10]。其一为支气管梗阻假说:STOCKER等[11]于1977年提出,MOERMAN等[12]所做的尸检为该假说提供了解剖证据,认为支气管功能性或器质性阻塞导致了肺发育过程中细胞增生和凋亡异常。其二为环境假说:基因缺陷或微环境变化导致调控因子异常表达而致使肺发育异常。肺发育的5个组织学阶段中,内胚层上皮细胞和间充质细胞的分子表达及信号通路起到关键作用[13],例如前肠腹侧上皮细胞的甲状腺转录因子(TTF1)的编码基因Nkx2-1、前肠背侧的性别决定区基因Y-box2、Sox2、Hoxb-5等;而BMPs和它的拮抗因子Noggin、FGF10、Wnt等则在间充质细胞内起作用[1]。当上皮细胞与间充质细胞之间的信号传导异常时,则导致肺实质内肺泡形成障碍,终末细支气管过度增生、扩张,形成单房、多房或蜂窝状错构瘤样的CPAM[1]。
本研究中,我们利用二代测序技术顺利得到了10个样本序列,质量均合格,顺利构建RNA-Seq文库。在做样本主成分分析时,发现样本3b和其他组内样品分隔开,为了顺利进行差异分析,该样品被排除在外。本研究共筛选出860个上调的基因和203个下调的基因,可见CPAM的发生与多个基因的上调和下调有关[1,13-15]。为明确上述差异表达基因集合的功能,我们采用R软件cluster Profiler与GO集合进行功能富集分析[9],包括分子功能、生物学过程和细胞组分三个部分。基因或蛋白质可以通过ID对应或者序列注释的方法找到与之对应的GO号,而GO号可对应到队列ID,即功能类别或者细胞定位。在GO功能富集分析结果中发现,细胞组分主要有轴丝、纤毛、微管、细胞顶端部分等,分子功能主要有微管蛋白结合、受体配体活性、信号受体激活、ATP酶活性、肌动活性、微管结合功能、G蛋白耦联受体结合、微管运动活性等,生物过程主要有纤毛组织、纤毛组装、轴丝组装、微管束形成、纤毛及鞭毛运动等。KEGG信号通路分析结果显示,差异表达基因参与介导了PI3K-Akt信号通路、血管平滑肌收缩、流体剪切应力和动脉粥样硬化、神经活性配体-受体相互作用、细胞因子-受体相互作用、亨廷顿病、转录失调、黏着斑、ECM-受体相互作用、病毒蛋白与细胞因子-受体相互作用、蛋白质消化吸收等多条信号通路,均涉及肺发育过程中的细胞增殖、迁移以及凋亡。通过GO功能富集分析和KEGG信号通路分析可预测CPAM的病理生理过程与纤毛及初级纤毛的形成、退化有关。
本研究基于二代高通量测序技术构建了稳定可靠的CPAM患儿RNA-Seq文库,筛选出差异表达基因1 063个:上调的基因860个,下调的基因203个。通过GO功能富集发现差异表达基因与纤毛形成、退化有关,KEGG信号通路分析提示差异表达基因参与了多个肺发育过程相关的信号通路。通过上述方法,初步明确各个基因的功能类别及细胞定位,预测了CPAM可能的发病机制。可见,生物信息学分析为研究CPAM转录调控网络及筛选分子生物学标志物提供了有效手段[21]。
(收稿日期:2021-06-23)
猜你喜欢
湘潮(上半月)(2022年7期)2022-12-06
自然杂志(2022年3期)2022-08-18
昆明医科大学学报(2022年2期)2022-03-29
中老年保健(2021年3期)2021-12-03
医学研究生学报(2021年4期)2021-12-02
猪业科学(2021年3期)2021-05-21
临床与实验病理学杂志(2021年3期)2021-04-25
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
世界农药(2019年2期)2019-07-13
