
超高效液相色谱-串联质谱法同时测定小麦粉中硫脲、曲酸、噻苯咪唑、噻二唑、四环素
2021-12-03 09:25:46李志刚赵文涛郭文萍李莹莹王娟强
食品科学 2021年22期
王 妍,李志刚,赵文涛,郭文萍,李莹莹,王娟强,姜 锐
(中国肉类食品综合研究中心,北京 100068)
小麦是我国三大农作物之一,其富含丰富的碳水化合物及蛋白质,主要用途为小麦粉的生产原料。国家统计局公布的数据显示,我国小麦产量逐年上升[1],小麦粉生产和消费快速增长,其安全和质量问题一度十分突出。在小麦粉加工、制作过程中,生产者为了满足小麦粉在感官性状、筋力、蛋白含量、稳定性等方面的要求,通常选择添加不同品种的优质小麦进行配粉,一些小作坊为了追求利益最大化,通过添加辅料或化学制剂的方式改善小麦粉的感官性状,掩盖原料本身质量不过关的事实;另外,小麦储存条件如不符合国家标准,在储藏过程中极易发生霉变或虫鼠危害导致小麦粉污染。不法商贩通过向小麦粉中添加增白剂、防腐剂和抗生素如硫脲[2-3]、曲酸、噻二唑[4]等非食品原料,使小麦粉达到出厂标准,这一行为会产生严重的食品安全风险,威胁消费者的身体健康[5-6]。为加强对小麦粉的监管,原国家食品药品监督管理总局发布第132号公告《总局关于进一步加强小麦粉质量安全监管的公告》,其中第四条“严禁生产企业在小麦粉中添加过氧化苯甲酰、次磷酸钠、硫脲、间苯二酚、过硫酸盐、噻二唑、曲酸等非食品原料。”明确禁止面粉中添加硫脲、曲酸、噻二唑等物质[7]。
目前,有关报道发现在小麦粉中检测出噻苯咪唑、四环素的药物残留,这严重影响了小麦粉的食品安全性。噻苯咪唑,是一种高效、低毒性的广谱杀菌剂,可用于小麦病害防治[8]。噻苯咪唑的长期接触或食入可能会导致免疫系统紊乱,严重威胁消费者健康。美国、日本等国家现已严格控制食品中该物质的残留量,我国也将食品中噻苯咪唑的残留量列为重要监测项目[9]。四环素是一种广谱杀菌药物,对革兰氏阳性菌、阴性菌、立克次体、滤过性病毒、螺旋体属乃至原虫类都有很好的抑制作用。对人体部分器官的细胞有很大的毒性,不仅会引起肠胃疾病,还会造成耐药细菌的出现[10]。
目前,鲜见文献报道同时测定小麦粉中这5种物质的检测方法。申科敏等[11]用高效液相色谱法同时测定小麦粉中曲酸、噻二唑、硫脲,方法检出限较高;目前食品中噻苯咪唑的检测方法主要有气相色谱法-串联质谱法[12]、液相色谱法[13-14]、液相色谱-串联质谱法[15-16]、表面增强拉曼法等[17];四环素常见的方法有液相色谱法[18]、液相色谱-串联质谱法[19-20],检测限均高于本方法。噻苯咪唑及四环素两种物质均鲜报道在面粉基质中的检测方法。本研究旨在采用超高效液相色谱-串联质谱技术,建立一种高通量、高灵敏度的快速定量检测方法,以期能够同时检测出小麦粉中硫脲、曲酸、噻二唑、噻苯咪唑、四环素5种物质,全面保障小麦粉的安全性。
1 材料与方法
1.1 材料与试剂
小麦粉 市售。
硫脲(纯度≥99.0%)、曲酸(纯度≥98.0%)上海安谱科技股份有限公司;噻苯咪唑(纯度≥98.9%)、四环素(纯度≥97.15%) 德国Dr.Ehrensorfer公司;噻二唑(纯度≥99.5%) 北京振翔科技有限公司;甲醇、乙腈、甲酸(均为色谱级),无水乙醇(分析纯) 国药集团化学试剂有限公司;水为一级水。
1.2 仪器与设备
1290高效液相色谱仪-6470串联四极杆质谱、涡旋振荡仪 美国Agilent公司;S-100涡旋仪 日本Taiyo公司;离心机 日本Hitachi公司;Milli-Q纯水仪美国Millipore公司;HSC-24B水浴氮吹仪 天津恒奥科技有限公司。
1.3 方法
1.3.1 提取溶液及流动相制备
体积分数0.1%的甲酸溶液:移取甲酸1 mL,加水稀释至1 L,混匀。
体积分数80%的乙醇溶液:量取无水乙醇400 mL,加水稀释至500 mL,混匀。
1.3.2 样品处理
称2 g样品,加入5 mL 80%乙醇溶液,涡旋2 min,用乙腈定容至10 mL。将样品管置于高速涡旋仪,2 000 r/min,涡旋15 min。12 000 r/min离心5 min,取5 mL上清液于刻度管中,40 ℃水浴中氮吹干。加入1 mL 0.1%甲酸溶液,涡旋复溶,过0.22 μm滤膜,待上机。
1.3.3 标准溶液的制备
硫脲标准储备液:称取硫脲标准品10 mg用10 mL水溶解并定容,配制成质量浓度为1 mg/mL硫脲标准储备液,4 ℃冷藏保存。
曲酸、噻二唑、四环素标准储备液:称取曲酸、噻二唑、四环素标准品10 mg,用10 mL甲醇溶解并定容,分别配制成质量浓度为1 mg/mL标准储备液,-18 ℃冷冻保存。
噻苯咪唑标准储备液:称取噻苯咪唑标准品10 mg,用100%甲醇溶解并定容,配制成质量浓度为100 μg/mL噻苯咪唑标准储备液,-18 ℃冷冻保存。
混合标准中间液:移取硫脲、曲酸、四环素标准储备液各1.00 mL,噻苯咪唑标准储备液100 μL,噻二唑标准储备液10 mL配制成混合标准工作液。其中硫脲、曲酸、四环素质量浓度为10 μg/mL、噻二唑质量浓度为100 μg/mL、噻苯咪唑质量浓度为100 ng/mL,4 ℃冷藏保存。
混合标准曲线的制备:配制基质标准曲线,分别称取6 份空白小麦粉样品,每份2 g,依次移取混合标准中间液20、50、100、200、500、1 000 μL添加到样品中,使样品充分吸收,参照1.3.2节前处理过程进行处理。标准曲线的最终质量浓度分别为(以硫脲为例)100、250、500、1 000、5 000 ng/mL。
1.3.4 超高效液相色谱-串联质谱条件
色谱条件:色谱柱:ZORBAX Eclipse Plus C18(2.1 mm×50 mm,1.8 μm);柱温30 ℃;进样体积2 μL;流动相:A为0.1%甲酸溶液,B为乙腈;流速0.30 mL/min。洗脱梯度见表1。
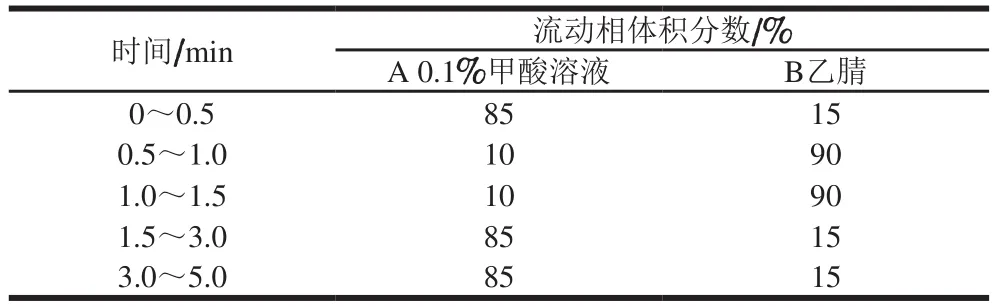
表1 梯度洗脱程序Table 1 Gradient elution procedure
质谱条件:电喷雾离子源(electron spray ionization,ESI);正模式喷雾电压3 000 V,负模式喷雾电压2 500 V,辅助气(氮气)气化温度300 ℃,鞘气(氮气)流速11 L/min,鞘气温度350 ℃。多反应监测质谱参数见表2。
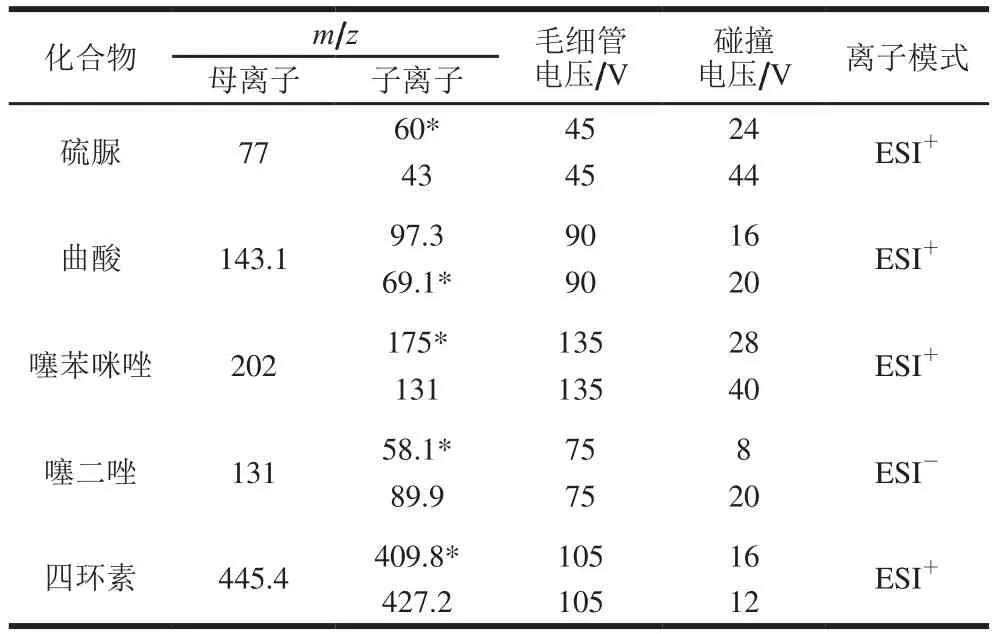
表2 5种物质多反应监测质谱参数Table 2 MRM mass spectrometric parameters for five substances
2 结果与分析
2.1 色谱条件优化
根据文献资料显示[21-25],5种物质在反向色谱柱C18柱上均有保留,故实验选取C18柱。
四环素容易在反相色谱柱的硅醇基上吸附,产生拖尾峰,需要在流动相中加入有机酸,才能取得较好分离度和峰形[26]。实验分别选取0.1%甲酸溶液-甲醇、(0.1%甲酸-5 mmol/L乙酸铵溶液)-甲醇、0.1%甲酸溶液-乙腈、(1%甲酸-5 mmol/L乙酸铵溶液)-乙腈4 种组合考察5种物质的保留及响应情况。结果表明,流动相含有乙酸铵时,负离子扫描模式下噻二唑的响应值较好,正离子扫描模式下的4 种物质的响应较低,曲酸和四环素的峰形较差。选择0.1%甲酸溶液作为水相,曲酸、硫脲、噻苯咪唑、四环素的响应及峰形较理想,噻二唑响应可满足实验要求。有机相为乙腈和甲醇对5种物质的峰形和响应的影响不明显,采用乙腈可以有效降低仪器系统内的压力,最终选择0.1%甲酸溶液-乙腈作为流动相。
如图1所示,5种物质因结构性的差异,与固定相之间产生的吸附力大小、强弱不同可以看出曲酸、硫脲极性较强,因此首先采用高比例的水相进行分离,硫脲和曲酸依次出峰。再加大有机相比例洗脱另外3 种物质,实现5种化合物较好的分离。
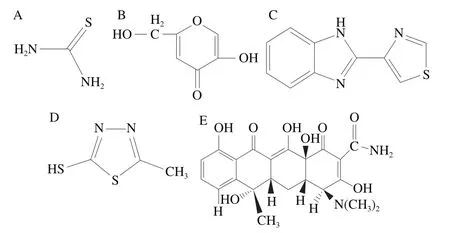
图1 5种物质的结构式Fig.1 Structural formulae of five substances
2.2 质谱条件的优化
硫脲结构中含有氨基,曲酸吡喃环上连接酚羟基和羟甲基,噻苯咪唑和四环素的分子结构也较容易得到H+形成[M+H]+型分子离子峰,因此硫脲、曲酸、四环素、噻苯咪唑均选择电喷雾离子正离子模式。噻二唑结构中含有一个巯基,易失去H+形成[M-H]-型分子离子峰,故化合物噻二唑选择电喷雾离子负离子模式。分别配制1 μg/mL的标准溶液,每个化合物在相应的模式下进行全扫描和单离子扫描得到母离子并优化碎裂电压,使母离子的丰度达到最优。随后进行子离子扫描,找到两个特征碎片离子,优化碰撞电压,并选取丰度较高的子离子作为定量离子。5种化合物的总离子流图、定量离子和定性离子图,见图2、3。
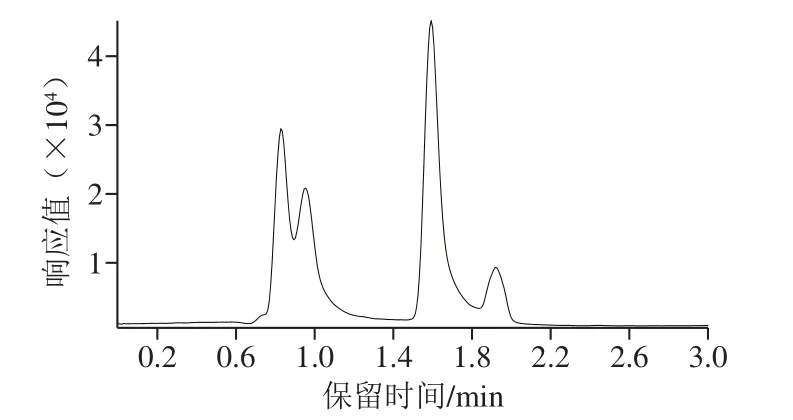
图2 5种化合物的总离子流图Fig.2 Total ion current chromatogram of five compounds
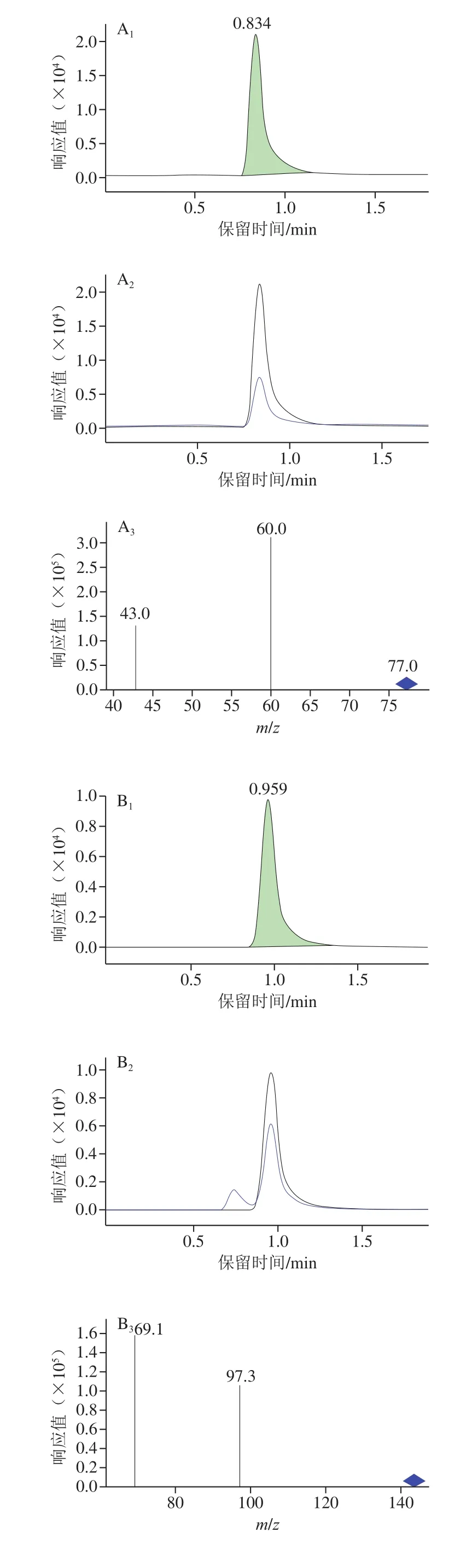
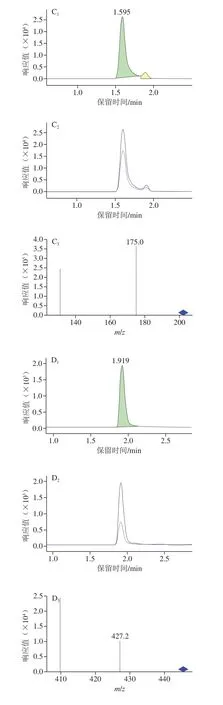

图3 5种物质的定量、定性及离子比例图Fig.3 Quantitative and qualitative chromatograms and mass spectra of five substances
2.3 前处理过程优化
根据5种物质的溶解性和相关文献资料的报道,实验选择甲醇、乙醇、乙腈、甲醇-乙腈(1∶1,V/V)、乙二胺四乙酸二钠-Mcllvsine缓冲液、水、80%乙醇-乙腈(1∶1,V/V)作为提取溶剂进行比较。选择乙二胺四乙酸二钠-Mcllvsine缓冲液、水提取样品并离心后,上清液较为浑浊,且仅有四环素回收率较好,其他化合物回收率均低于70%;甲醇、乙醇、乙腈、甲醇-乙腈(1∶1,V/V)、80%乙醇-乙腈(1∶1,V/V)沉淀蛋白效果较好,采用氮吹方式除杂和浓缩后回收率可得到较优结果。
实验结果表明,添加100 μL混合标准中间液(1.3.1.2节),相当于硫脲、曲酸、四环素添加量为250 μg/kg,噻苯咪唑为2.5 μg/kg,噻二唑为2.5mg/kg时,采用80%乙醇溶液-乙腈(1∶1,V/V)混合体系为提取溶剂时,5 种物质的回收率可达到80%以上。综上,本研究最终选择80%乙醇溶液-乙腈(1∶1,V/V)作为提取溶剂,并采用氮吹方式除杂和浓缩。不同提取溶剂对回收率的影响见图4。

图4 提取溶剂对回收率的影响Fig.4 Effect of different extraction solvents on recoveries of five analytes
本实验还考察不同复溶溶液对5 种物质回收率的影响。向空白样品基质溶液中加入标准溶液后吹干,分别用1 mL 0.1%甲酸溶液、0.1%甲酸溶液-乙腈(1∶1,V/V)、乙腈定容,涡旋混合后过0.22 μm滤膜上机测定。添加100 μL混合标准中间液(1.3.3节),相当于硫脲、曲酸、四环素添加量为250 μg/kg,噻苯咪唑为2.5 μg/kg,噻二唑为2.5mg/kg时,结果见图5。0.1%甲酸溶液复溶样品,5 种物质回收率较好,干扰小,峰形、响应均较为理想,故本实验选取0.1%甲酸溶液作为复溶溶剂。
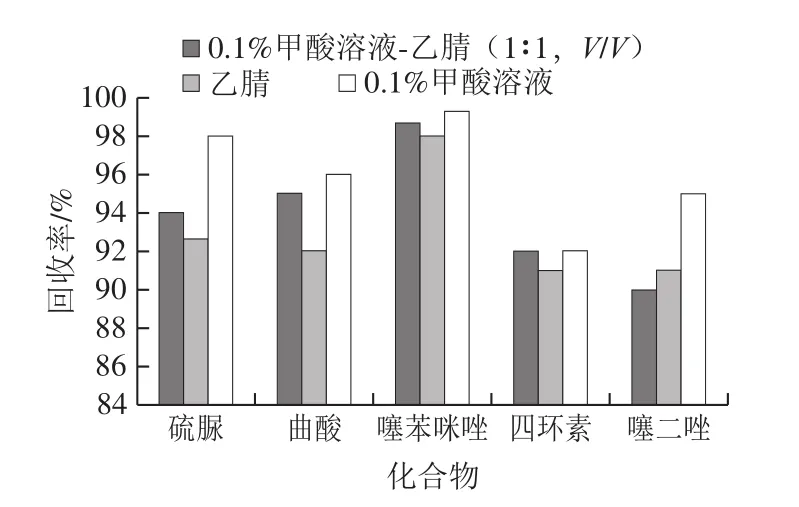
图5 复溶溶液对5 种物质回收率的影响Fig.5 Effect of different redissolution solvents on recoveries of five analytes
2.4 方法学结果
2.4.1 基质效应
样品经前处理后依然保留在待测液中的糖类、可溶性蛋白或肽类等有机化合物、无机盐等杂质与目标化合物共流出,进入电离源,影响目标化合物的离子化效率,即基质效应[27]。引起目标化合物响应降低称为基质抑制效应,响应升高为基质增强效应[28]。本研究通过提取后添加法评价5 种待测物在小麦粉中的基质效应[29]。将空白样品按前处理步骤提取净化后,向提取液中添加不同质量浓度的标准溶液,制作基质匹配标准溶液。另外制作一条用流动相配制的相同质量浓度梯度的溶剂标准溶液[30-31]。分别测定并根据质量浓度与峰面积绘制曲线得到两条曲线斜率,根据下式计算基质效应[32]:
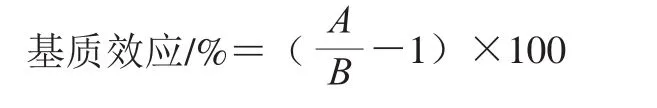
式中:A为基质匹配标准曲线斜率;B为溶剂标准曲线斜率。
实验结果显示,小麦粉基质对噻苯咪唑产生的基质效应不明显,对噻二唑、四环素、曲酸、硫脲的测定存在基质抑制效应,见表3。有研究指出,处理生物样品时不采用空白基质配制标准溶液法,而应采用空白基质标准前加标法消除或补偿基质效应[34]。故本研究采用空白基质前加标法制作标准曲线,有效消除基质效应,降低硫脲、曲酸回收率低对结果准确性的影响[35]。
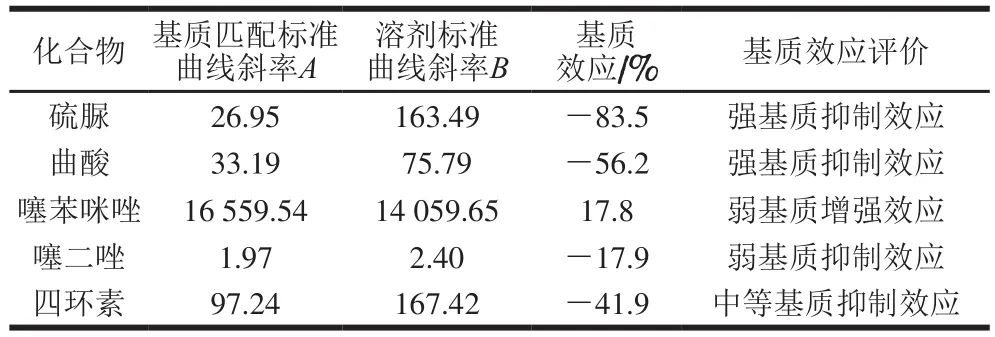
表3 小麦粉中5 种物质的基质效应评价Table 3 Matrix effect evaluation of five analytes in flour
2.4.2 线性、检出限与定量限
称取6 份空白小麦粉样品,每份2 g,依次移取混合标准中间液20、50、100、200、500、1 000 μL添加到样品中,按1.3.2节样品处理过程操作,制作基质加标曲线。分别绘制线性方程,硫脲、曲酸、四环素线性范围为100~5 000 ng/mL,噻苯咪唑线性范围为1~50 ng/mL,噻二唑线性范围为1~50 μg/mL,相关系数在0.999以上,线性关系良好,见表4。硫脲、曲酸、四环素检出限为25 μg/kg,定量限为50 μg/kg。噻苯咪唑检出限为0.5 μg/kg,定量限为1 μg/kg。噻二唑检出限为0.5 mg/kg,定量限为1 mg/kg。检出限满足RSN≥3,定量限满足RSN≥10。
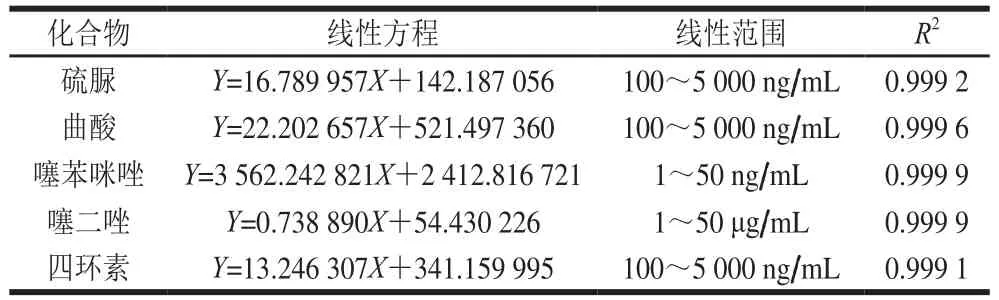
表4 小麦粉中5 种物质的线性关系Table 4 Calibration curve equations of five analytes in flour
2.4.3 回收率与精密度实验
选取空白小麦粉样品,每份2 g,分别添加低、中、高水平标准溶液,每个添加水平做6 次平行实验,结果见表5。结果表明3 个不同添加水平的回收率在88.6%~102.2%之间,相对标准偏差在0.7%~5.6%之间,均符合GB/T 27404—2008《实验室质量控制规范食品理化检测》中要求。
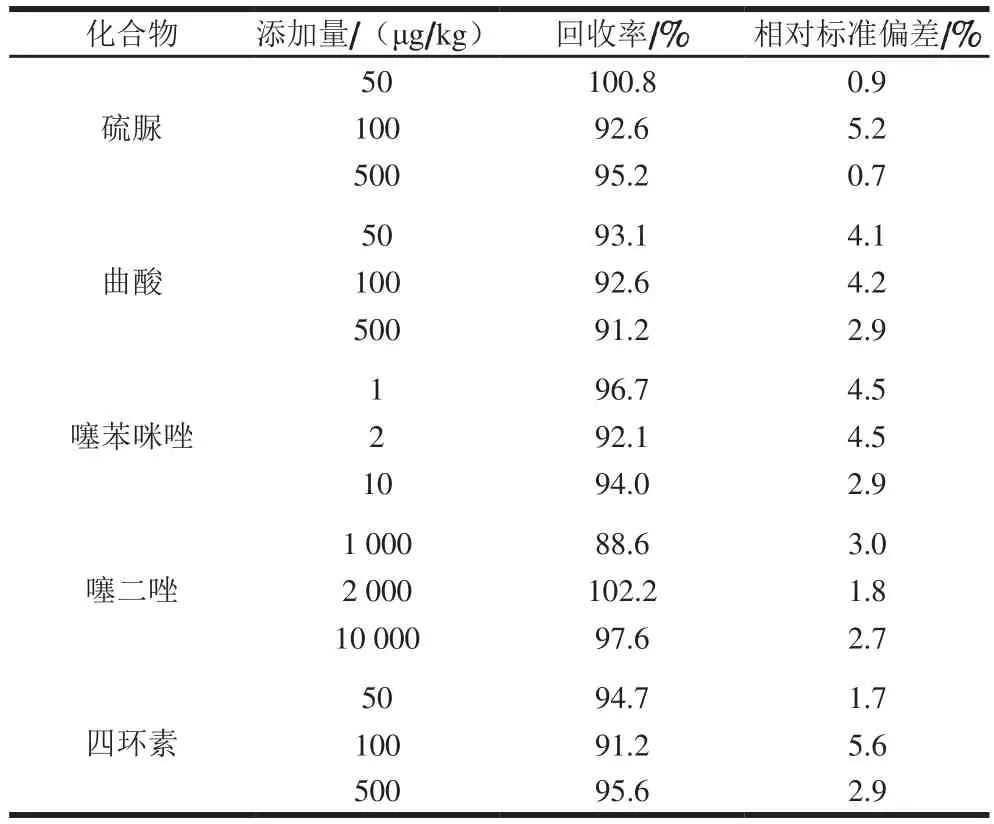
表5 小麦粉基质中5 种化合物不同添加水平回收率结果Table 5 Recoveries of five analytes in flour matrix at different spiked levels
2.5 实际样品的测定
按照本方法对市售25 批次小麦粉进行分析,其中1 批次检测出噻苯咪唑残留。本方法可用于实际样品的快速定量检测。
3 结 论
本实验建立了一种利用超高效液相色谱-串联质谱技术同时测定小麦粉中硫脲、曲酸、噻苯咪唑、噻二唑、四环素物质的检测方法,其中硫脲、曲酸、四环素检出限为25 μg/kg,定量限为50 μg/kg;噻苯咪唑检出限为0.5 μg/kg,定量限为1 μg/kg;噻二唑检出限为0.5 mg/kg,定量限为1 mg/kg。方法灵敏度、回收率高、重复性好、前处理操作简便快速,在实际检测中具有较强的可操作性,可以作为小麦粉中非法添加物的定量检测方法。
猜你喜欢
化工管理(2021年7期)2021-05-13 00:45:38
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19 08:38:52
中国棉花(2021年5期)2021-01-02 02:37:52
农药科学与管理(2019年8期)2019-11-23 08:04:44
兴义民族师范学院学报(2018年5期)2018-12-18 03:16:54
红外技术(2017年1期)2017-03-27 09:02:21
广州城市职业学院学报(2016年2期)2016-07-25 07:39:30
新疆农垦科技(2015年11期)2015-09-08 10:15:39
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:19
新疆农垦科技(2014年7期)2014-02-28 19:20:29
