
Genome-wide map of N6 -methyladenosine circular RNAs identified in mice model of severe acute pancreatitis
2021-12-03 06:15:34JunWuXiaoHuiYuanWenJiangYiChenLuQiLinHuangYiYangHuaJiQieJiangTaoLiuHongYuSunLiJunTang
World Journal of Gastroenterology 2021年43期
Jun Wu, Xiao-Hui Yuan, Wen Jiang, Yi-Chen Lu, Qi-Lin Huang, Yi Yang, Hua-Ji Qie, Jiang-Tao Liu, Hong-Yu Sun, Li-Jun Tang
Abstract
Key Words: Severe acute pancreatitis; Circular RNAs; N6 -methyladenosine; MeRIP-seq;Epigenetic analysis
INTRODUCTION
Acute pancreatitis (AP) is a pancreatic inflammatory disorder that is associated with substantial morbidity and mortality[1]. Approximately 20 % of patients with AP develop into severe AP (SAP)[2]. Due to the extensive pancreatic necrosis, subsequent infection, systemic inflammatory response syndrome and multiple organ failure, the mortality of SAP is up to 30 %[2 ,3]. Previous studies have suggested that some important pathological mechanisms, including premature trypsinogen activation in the acinar cells and macrophages, mitochondrial dysfunction, pathological calcium signaling, endoplasmic reticulum (ER) stress, and impaired autophagy, are involved in the initiation and development of SAP[1]. However, the pathophysiology of SAP is complex and remains unclear, especially the level of gene regulation.
CircRNAs were discovered in the 1970 s[4] and were identified as single-stranded covalently closed RNA molecules that lack 5 ’ caps and 3 ’ tails[5]. Long after, they were thought to be the byproducts of splicing[6]. In recent years, as high-throughput sequencing developed, thousands of circRNAs were found to be expressed in a wide range of mammalian tissues[7 ,8], including the pancreas[9], and accumulating studies have demonstrated that circRNAs play vital roles in the whole process and prognosis of many diseases, including cardiovascular diseases[8], cancer[10], neurodevelopmental processes[11], immune responses and immune diseases[12]. The main mechanisms of circRNAs participated in the initiation and development of diseases include the following functions[6 ,8 ,10 ,12]: interplay with RNA-binding proteins,microRNA (miRNA) sponges, regulating the stability of mRNAs, modulating the transcription of parental gene and the templates for protein synthesis. However, the post-transcription modification of circRNAs remains unclear.
N6-methyladenosine (m6 A) is the most prevalent internal modification of RNA in eukaryotic cells[13]. In 2017 , Zhou et al[14] reported that the m6 A modification is widespread in circRNAs and m6A modifications are read and written by the same complexes in circRNAs and mRNAs. The regulatory role of m6A is mainly performed by three homologous factors, namely so-called “writers”, “erasers” and “readers”[13 -15]. The writers mainly include methyltransferase-like 3 and 14 proteins (METTL3 and METTL14 ) and their cofactor WT1 -associated protein (WTAP). They form a methyltransferase complex to catalyze the installation of m6A. The erasers, including alkylation repair homolog 5 (ALKBH5 ) and fat mass and obesity related protein (FTO),can catalyze the oxidative demethylation of N-alkylated nucleic acid bases. The readers are mainly YT521 -B homology (YTH) domain containing proteins family,including YTHDC1 , YTHDC2 , YTHDF1 , YTHDF2 and YTHDF3 . They can specifically recognize m6A and regulate splicing, localization, degradation and translation of RNAs. Recently, it has been found that the m6A modification of circRNAs plays a key role in innate immunity and tumors though regulating the metabolism and function of circRNAs[15]. In human embryonic stem cells and HeLa cells, m6 A circRNAs display cell-type-specific methylation patterns[14]. In colorectal carcinoma, the m6 A modification can modulate cytoplasmic export of circNSUN2 and stabilize HMGA2 ,ultimately enhancing the colorectal liver metastasis[16]. However, the roles of m6 A circRNAs in SAP are still unknown.
Here, we investigated the expression profile of m6A circRNAs in SAP through m6 Amodified RNA immunoprecipitation sequencing (MeRIP-seq). We evaluated the biological significance of circRNAs with differentially expressed m6A peaks though gene ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG)pathway analysis, and explored their underlying mechanism by construction of m6A circRNA-miRNA networks. In addition, we determined the expression of demethyltransferase, ALKBH5 and FTO, to deduce the possible mechanism of reversible m6 A process in SAP.
MATERIALS AND METHODS
Animals and preparation of SAP model
Male C57 BL/6 mice weighing 22 -25 g were provided by Chengdu Dashuo Experimental Animal Technology Co. Ltd. All the mice were housed in ventilated plastic cage system and fed with the same food and water for 7 d to adapt to the environment.The entire research protocol was approved by the Institutional Animal Care and Use Committee at the General Hospital of Western Theater Command.
Before the operation, the mice were divided into SAP and control groups randomly(3 mice per group) and fasted for 12 h but had free access to water. Isoflurane (5 %) was used to anesthetize mice by induction box prior to surgery. Then, the SAP was induced through 4 % sodium taurocholate salt that was slowly retrogradely injected into the biliopancreatic duct with a microinfusion pump. All mice were killed 24 h after the establishment of model, and the blood samples and pancreatic tissues were collected for further analysis.
Pancreatic histological analysis
Pancreatic tissue (0 .4 cm × 0 .4 cm) was fixed in 4 % paraformaldehyde solution. After dehydrating with ethanol, the tissue samples were embedded in paraffin. Then, the samples were cut into about 4 -μm-thick sections, and the sections were stained with hematoxylin and eosin. The light microscopy at × 200 magnification was used to examine the slide. The scoring system described previously was used to evaluate the degree of pancreatic injury[17]. The scores were averaged for five different slides that were selected randomly from each pancreas.
Amylase and lipase measurement
The concentrations of lipase and amylase in serum were determined using Lipase Assay kit and Amylase Assay kit (Nanjing Jiancheng Bioengineering Institute,Nanjing, China) according to the instructions.
RNA isolation and RNA quality control
TRIzol reagent (Invitrogen, Carlsbad, CA, United States) was used to extract total RNA from the homogenized pancreatic tissues of the control and SAP groups. The concentration of extracted RNA was measured at OD260 and 280 by NanoDrop ND-2000 instrument (Thermo Fisher Scientific, Waltham, MA, United States). We assessed the integrity of RNA through denaturing agarose gel electrophoresis. The OD A260 /A280 ratio between 1 .8 and 2 .0 was set as the RNA purity standard.
Library preparation and MeRIP-seq
rRNAs in total RNA were removed using Ribo-Zero rRNA Removal Kits (Illumina,San Diego, CA, United States). The removal efficiency of rRNA by the residual determination of 28 S and 18 S of rRNA using quantitative polymerase chain reaction (qPCR).The fragmented RNA was incubated with the anti-m6A antibody at 4 °C for 2 h in IPP buffer. Then, the mixture was immunoprecipitated by incubation with protein-A beads (Thermo Fisher Scientific) for 2 h at 4 °C. The bound RNA was eluted from the beads with m6A (Berry & Associates) in IPP buffer and then extracted with TRIzol reagent (Thermo Fisher Scientific). The immunoprecipitated RNA and input RNA were used to construct the library using NEBNext®Ultra™ RNA Library Prep Kit and double-ended 150 -bp sequencing of the m6 A-IP and input samples was performed on an Illumina HiSeq sequencer (performed by Cloudseq Biotech Inc., Shanghai, China).
Analysis of MeRIP-Seq data
Paired-end reads were harvested from the Illumina HiSeq 4000 sequencer, and were quality controlled by Q30 . To obtain high quality clean reads, 3 ’ adaptor-trimming and low-quality reads were removed by cutadapt software. The clean reads with high quality of the input library were aligned to the mouse reference genome (UCSC MM10 ) with STAR software. DCC software was used for detecting and identifying the circRNAs. The identified circRNAs were annotated using the circBase database and Circ2 Traits database. For all samples, raw junction reads were normalized to the number of total mapped reads and log2 transformed. The read alignments on the genome were visualized using the tool integrative genomics viewer. The adapterremoval reads were aligned to the reference genome using Hisat2 software. The methylated sites in each sample were identified using MACS software. Differentially methylated sites were identified using diffReps software.
GO and KEGG analysis
The parent genes of circRNAs with differential m6A peaks were selected to analyze their potential biological roles through GO and KEGG pathway analysis. GO analysis included three parts, namely, biological process (BP) analysis, molecular function (MF)analysis, and cell component (CC) analysis[18]. GO analysis was performed by R topGO package. Fisher’s exact test in Matlab MCR software was applied to calculate the enrichment of each pathway. The bubble plots and column plots were generated using the ggplot2 in R package (https://ggplot2 .tidyverse.org).
Construction of circRNA-miRNA networks
circRNA containing miRNA-binding sites can bind to miRNA response elements competitively, further regulating the target mRNAs[19]. The top 10 upregulated and top 10 downregulated circRNAs according to the level of m6 A were selected to construct circRNA-miRNA networks. The m6A circRNA-miRNA networks were constructed using TargetScan software and miRanda software and the circRNA-miRNA interactions were visualized by Cytoscape.
Conservation analysis
The top 10 upregulated and top 10 downregulated circRNAs were selected to analyze their homology with human circRNAs. The sequence of human circRNAs was downloaded from circBase database and the sequence of each selected m6A circRNA was blasted against the human circRNAs sequence by the blastn function of Blast software.
Western blotting
The whole pancreatic tissues from SAP and control groups were placed in RIPA lysate buffer with protease inhibitor, phosphatase inhibitor and phenylmethylsulfonyl fluoride inside (Total Protein Extraction Kit; Beijing Solarbio Science and Technology Inc., Beijing, China), and the tissues were homogenized with homogenizer. The tissue homogenate was centrifuged at 12000 g for 30 min at 4 °C, and the supernatant was collected. After protein concentration was measured by BCA Protein Assay Kit(Beyotime Biotechnology, Jiangsu, China), the supernatant was mixed with loading buffer (Beijing Solarbio Science and Technology), boiled at 100 °C for 10 min for protein denaturation, and stored at -80 °C after separation. The target proteins were separated by SDS-PAGE. The proteins were transferred to polyvinylidene difluoride membrane (0 .45 μm, IPVH00010 ; Millipore, Billerica, MA, United States), blocked in 5 % nonfat milk for 1 h at room temperature (22 ± 3 °C), and then incubated with primary antibody, FTO (1 :1000 , D2 V1 I; Cell Signaling Technology, Danvers, MA,United States), ALKBH5 (1 :2000 , 16837 -1 -AP; Proteintech, Rosemont, IL, United States), GAPDH (1 :5000 , 10494 -1 -AP; Proteintech) at 4 °C overnight. The membranes were washed with Tris-buffered saline with Tween-20 (TBST) (Beijing Solarbio Science and Technology) three times and incubated with secondary antibody (1 :10000 , 15015 ;Proteintech) at room temperature for 1 h. After being washed three times with TBST,the protein bands were visualized by enhanced chemiluminescence (Immobilon Western Chemilum HRP Substrate; Millipore) in a biological imaging system.
qPCR
The total RNA was extracted from SAP and control groups as described above. qPCR was performed using One Step SYBR®PrimeScript™ RT-PCR kit II (Takara Biotechnology Co., Ltd., Dalian, China) and the primers (ALKBH5 : forward 5 ’-GGCGGTCATCATTCTCAGGAAGAC-3 ’ and reverse 5 ’-CTGACAGGCGATCTGAAGCATAGC-3 ’;FTO: forward 5 ’-CTCACAGCC TCGGTTTAGTTCCAC-3 ’ and reverse 5 ’-CGTCGCCATCGTCTGAGTCATT G-3 ’; GAPDH: forward 5 ’-GGTGAAGGTCGGTGTGAACG-3 ’ and reverse 5 ’-CTCGCTCCTGGAAGATGGTG-3 ’) were synthesized by Shanghai Sangon Biotech Co., Ltd.. The outcomes were analyzed by means of 2-ΔΔCTthrough normalizing the quantity of GAPDH.
Data analysis
GraphPad Prism 8 (La Jolla, CA, United States) and SPSS 22 .0 (IBM Corp., Armonk,NY, United States) were used for performing statistical analyses. Student’sttest was used for estimating statistically significance between two groups. The results were evaluated through Spearman’s correlation coefficient test. All values are shown as mean ± SE of the mean;P< 0 .05 was regarded as statistically significant.
RESULTS
Evaluation of mouse model of SAP
Twenty-four hours after treatment with sodium taurocholate salt, the staining of hematoxylin and eosin on the pancreatic tissues from the SAP group showed typical histopathological changes, including pancreatic lobular edema, extensive acinar cell necrosis, focal expansion of the pancreatic interlobular septum and granulocyte infiltration (Figure 1 A). By contrast, under light microscopy, the pancreases from the control group had a complete normal structure. Figure 1 B showed the corresponding histopathological scores. At the same time, considering that the levels of serum lipase and amylase are as one of the diagnostic criteria of AP[20], we determined their concentrations in serum. As a result, the serum lipase and amylase levels in the SAP group were also markedly higher than those in the control group (P< 0 .05 ; Figure 1 C and 1 D). These results confirmed the successful establishment of the SAP mice model.
Overview of m6 A circRNAs in SAP
We used MeRIP-seq to investigate the expression of m6A circRNAs in pancreatic tissues from the control and SAP groups. We had submitted the data to the online repository, which can be found at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE173298 . Before performing MeRIP-seq, the residual determination of 28 S and 18 S of rRNA showed that the rRNAs in total RNA were removed effectively(Supplementary Figure 1 ). In general, a total of 409 m6 A circRNAs were identified in all chromosomes (Figure 2 A). Among these, 178 were specifically expressed in the SAP group, 107 in the control group, and 124 were shared in both groups (Figure 2 B). m6 A level in total circRNAs from the SAP group was lower than that from the control group (Figure 2 C). Besides, > 80 % of circRNAs contained only one m6 A peak in both SAP and control groups (Figure 2 D).
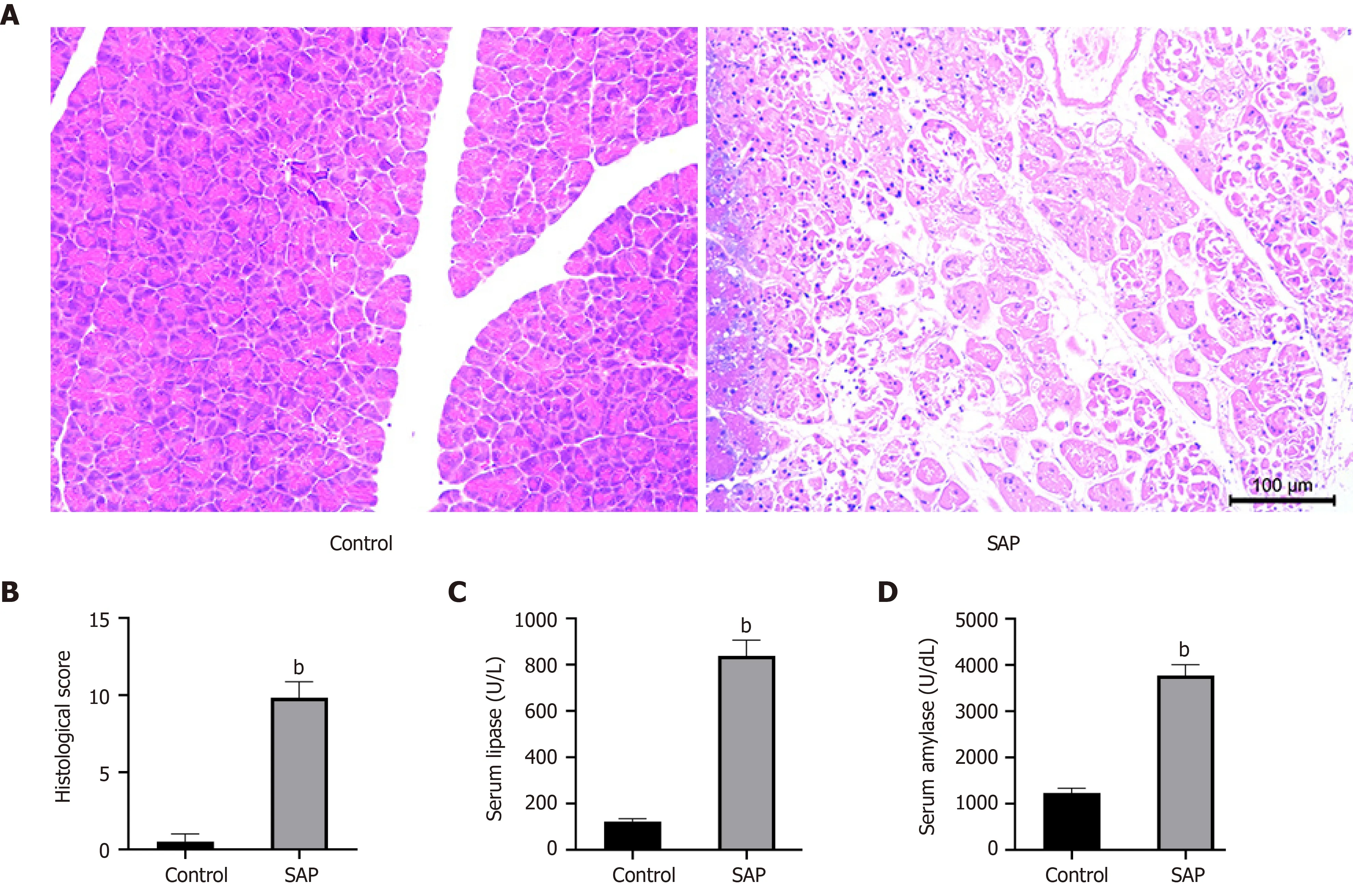
Figure 1 Evaluation of mouse model of severe acute pancreatitis. A: Representative images of pancreatic tissues stained with hematoxylin from control(left) and severe acute pancreatitis (SAP) (right) groups (× 100 magnification); B: Histological score of pancreatic tissues in control and SAP groups; C and D: Levels of serum lipase and amylase, respectively. bP < 0 .01 vs control group, n = 3 per group.
Differential m6 A modification of circRNAs in SAP
To understand the biological role of m6A modification of circRNAs in SAP, the circRNAs with differentially expressed (DE) m6A peaks were further analyzed.Significant differential expression was defined as fold-change > 2 and P < 0 .05 .Compared with the control group, 57 circRNAs with DE m6 A peaks were identified; 32 were upregulated and 25 downregulated in the SAP group. Table 1 presents the top 10 methylated m6A sites that were up- and downregulated within circRNAs. Figure 3 A shows the m6A circRNAs expression profile in the SAP and control groups though hierarchical cluster analysis. The scatter plot exhibits the variation of DE m6A circRNAs between the SAP and control groups (Figure 3 B). The volcano plot depicted DE m6A circRNAs between the two groups (Figure 3 C).
Distribution of m6 A sites in SAP and control groups
We identified 903 m6 A peaks distributed on 781 circRNAs and it is reported that circRNAs can be generated from any region of the genome[21]. Therefore, we firstly analyzed the genomic distribution of m6A and non-m6 A circRNAs according to their genomic origins to explore their distribution features. As a results, in non-m6A circRNAs, 45 .33 % were sense overlapping, 21 .15 % exonic, 26 .71 % intronic, 4 .94 %intergenic and a few antisense; in m6A circRNAs, 42 .78 % were sense overlapping,30 .32 % exonic, 21 .27 % intronic, 3 .42 % intergenic and a few antisense (Figure 4 A).These results indicated that the majority of m6A and non-m6 A circRNAs were commonly encoded by sense overlapping sequences and the number of circRNAs that generated from protein-coding genes in m6A circRNAs was more than those in non-m6 A circRNAs.
We further analyzed the distribution of circRNAs with DE m6A peaks. The length of DE m6A circRNAs was mainly enriched in 1 -10000 base pairs (Figure 4 B). Although the host genes of m6A circRNAs located in all chromosomes, the dysregulated parts mostly located in chromosomes 4 , 9 and 11 (Figure 4 C). A previous study reported that most circRNAs that derived from protein-coding genes spanned two or three exons[14]. In this study, the majority of circRNAs from protein-coding genes spanned one or two exons (Figure 4 D). Similarly, the majority of m6 A circRNAs and non-m6 AcircRNAs were more commonly encoded by a single or two exons (Figure 4 E).
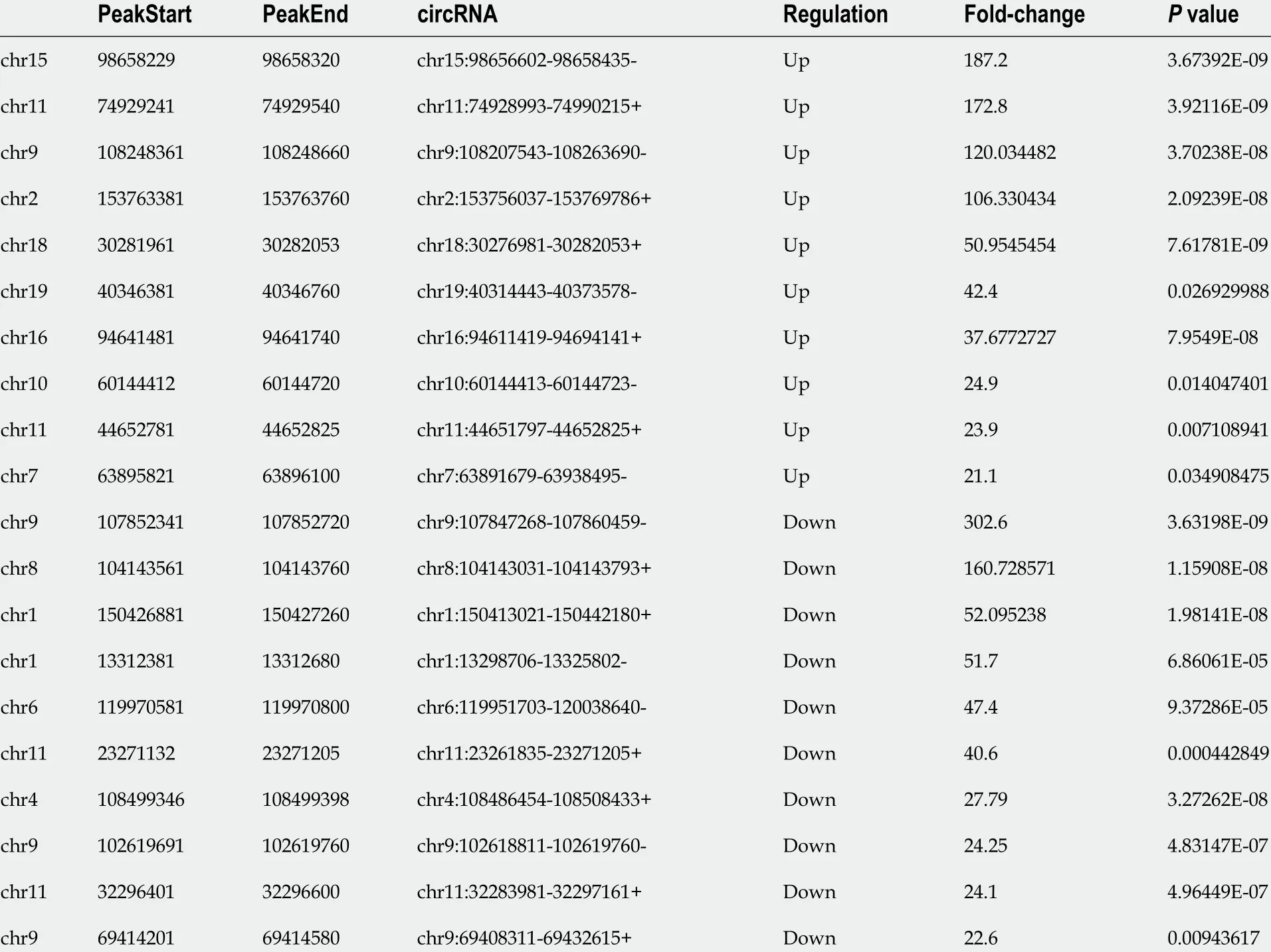
Table 1 Top 20 differently expressed N6 -methyladenosine peaks compared with control group
Functional analysis of circRNAs with DE m6 A peaks
To explore the function of m6A circRNAs in SAP, GO analysis and KEGG pathway analysis of circRNAs with the DE m6A peaks were performed. Figure 5 A presented the top 10 GO terms of circRNAs with upregulated m6 A peaks from the three aspects: BP,CC and MF. For BP, the most enriched and meaningful GO terms were cellular component organization, macromolecule metabolic process and regulation of developmental process. For CC, the top three terms were focal adhesion, cell-substrate junction and anchoring junction. For MF, the main represented GO terms were C2 H2 zinc finger domain binding and protein binding. The top 10 pathways from KEGG pathway analysis for circRNAs with upregulated m6A peaks were selected and presented in a bubble chart (Figure 5 B). Among them, protein digestion and absorption and regulation of autophagy were the major signaling pathways associated with the SAP progression.
The GO terms of circRNAs with downregulated m6A peaks are presented in Figure 5 C. For BP, protein-containing complex localization, RNA transport and macromolecule metabolic process were the most enriched and meaningful GO terms.For CC, nucleus, dendrite and dendritic tree were the top three terms. For MF, the main represented GO terms were channel regulator activity, RNA, enzyme and protein binding. As for the KEGG pathway analysis of circRNAs with downregulated m6A peaks, RNA transport was the main pathway (Figure 5 D).
Relationship between m6 A level and expression of circRNAs in SAP
To explore whether m6A modification could affect the expression of circRNAs, we analyzed the expression of m6A circRNAs. The expression level of these circRNAs with DE m6A peaks did not have significant differences (fold-change < 2 or P > 0 .05 ;Supplementary Table 1 ), indicating that m6 A modification of circRNAs did not influence the expression of circRNAs. To verify this result further, we analyzed the cumulative distribution of circRNA expression between the control and SAP groups for m6A and non-m6 A circRNAs (Figure 6 ). This was consistent with the above result.
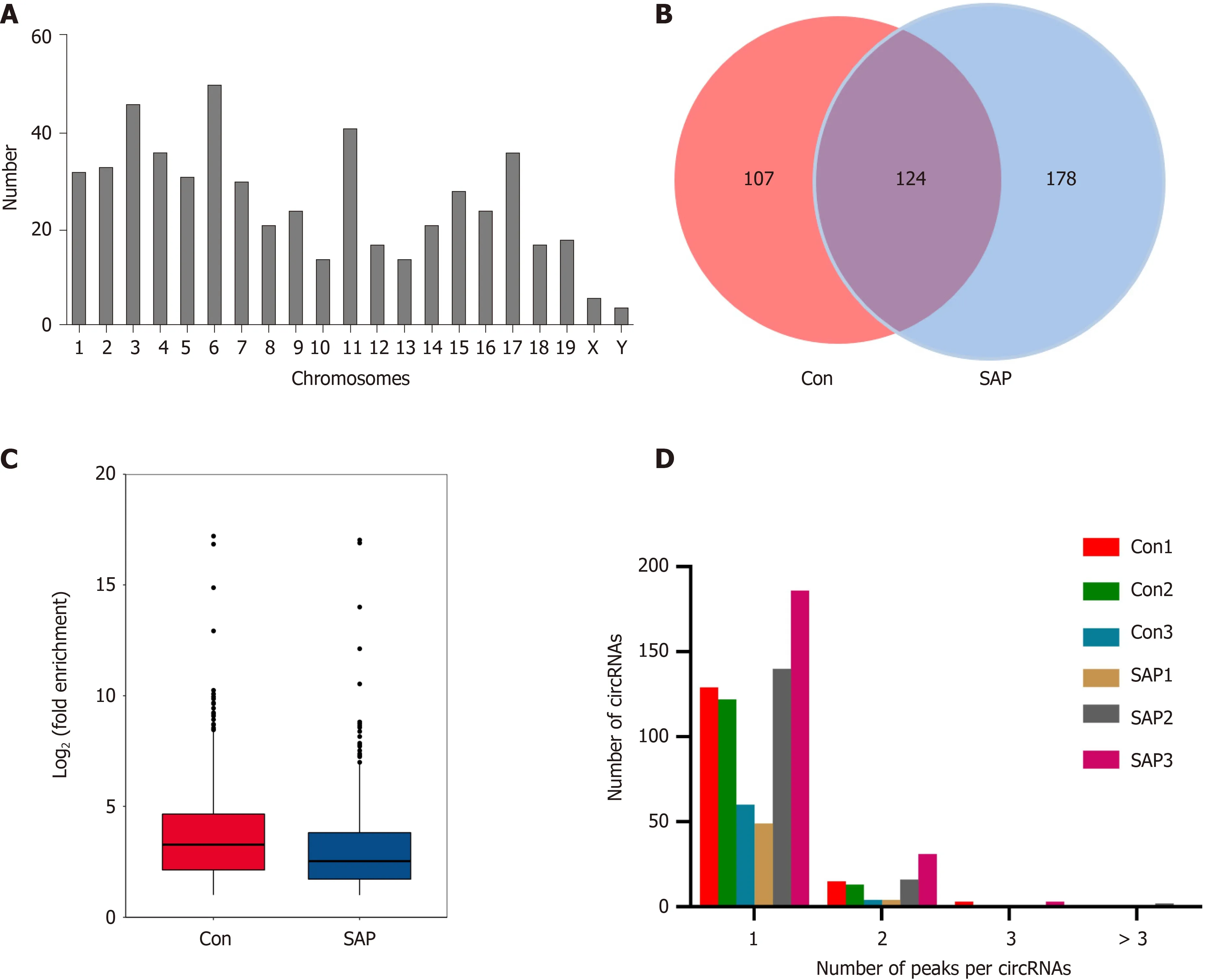
Figure 2 Overview of N6 -methyladenosine circRNAs in severe acute pancreatitis. A: Number of identified N6 -methyladenosine (m6 A) circRNAs according to distribution on chromosomes; B: Venn diagram exhibiting number of common and specific m6 A circRNAs between control and severe acute pancreatitis(SAP) groups; C: Box plot showing level of m6 A peaks enrichment in circRNAs in control and SAP groups; D: Number of circRNAs containing variant numbers of m6 A peaks.
Construction of m6 A circRNA-miRNA networks in SAP
Given the importance of circRNA-miRNA interaction[22] and to further explore the underlying mechanism of these circRNAs with DE m6A peaks, the top 10 upregulated and top 10 downregulated circRNAs according to the level of m6 A were selected to construct circRNA-miRNA networks. In this network map, several important miRNAs participated in the occurrence and development of SAP were found to bind to these m6A circRNAs (Figure 7 ), such as miR-24 -3 p, miR-26 a, miR-92 b, miR-216 b,miR-324 -5 p and miR-762 . These data suggest that these circRNAs with DE m6 A peaks might play a role in the pathological process of SAP.
Conservation analysis of identified m6 A circRNAs with human circRNAs
To explore whether the circRNAs with DE m6A peaks identified in mouse SAP may have similar roles in human SAP, we performed the conservation analysis of the sequence of the top 10 upregulated and top 10 downregulated circRNAs preliminarily.Through aligning with the sequence of human circRNAs that downloaded from circBase database, we found that 15 /20 of the selected circRNAs that have highly similar sequences to human circRNAs (sequence identity > 80 %), as shown in the Table 2 . These results suggested that these circRNAs may have similar roles in human SAP.
Expression of demethyltransferase in SAP
Given that the total m6A level of circRNAs was reduced in SAP and to explore how the m6A level was regulated in SAP, we detected the protein and mRNA expression of twodemethyltransferases (ALKBH5 and FTO). FTO was reduced at the level of protein,but ALKBH5 was increased in SAP at both the level of mRNA and protein (Figure 8 ).These results indicated that ALKBH5 might be related to the dynamic process of m6 A in SAP.
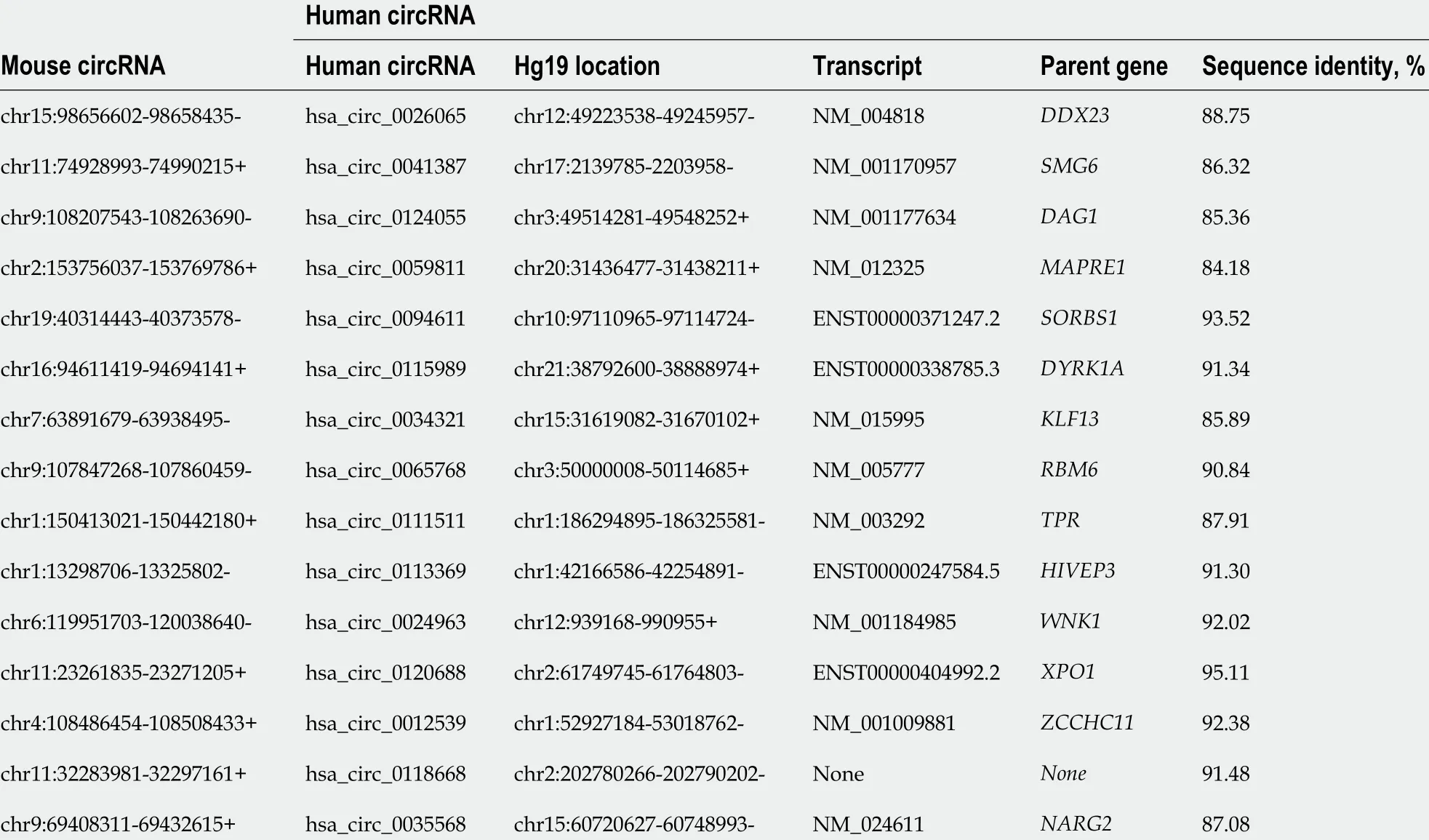
Table 2 The conservation analysis of the sequence between the selected circRNAs and human circRNAs
DISCUSSION
In the present study, we identified transcriptome-wide map of m6A circRNAs and determined their biological significance and potential mechanisms for the first time in SAP. The main findings are: (1 ) We identified 57 circRNAs with DE m6 A peaks and found these DE m6A circRNAs were involved in the key process of SAP by GO and KEGG analysis, such as protein digestion and regulation of autophagy; (2 ) In m6 A circRNA-miRNA networks, several important miRNAs participated in the initiation and development of SAP were found to bind to these m6A circRNAs potentially,suggesting that m6A may affect the interplays with miRNAs; and (3 ) The total m6 A level was reduced in SAP, and the demethylase ALKBH5 was found to be upregulated in SAP, indicating that ALKBH5 may be related to dynamic process of m6 A in SAP.These results suggested that m6A modification on circRNAs may be involved in the pathophysiology of SAP, which may provide novel insights to explore the possible pathophysiological mechanism of SAP and seek new potential therapeutic targets.
To find effective therapeutic targets for SAP, many studies have explored the underlying molecular mechanisms of SAP. Our previous study found that many circRNAs are expressed in mice with SAP[9] and these circRNAs play an important role in the pathogenetic mechanism of SAP[9 ,23]. In recent years, m6 A modification of circRNAs was found to be widespread[14] and gained widespread attention in epigenetics. Several important studies have investigated the roles of m6A modification in circRNA metabolism and found that m6A circRNAs play key roles in some diseases[16 ,24 -28]. In circRNA metabolism, m6 A modifications can regulate its translation through recognition by YTHDF3 and eIF4 G2 , and this progress of translation can be enhanced by METTL3 /14 and inhibited by FTO[24 ,25]. In addition, m6 A circRNAs associate with YTHDF2 in an HRSP12 -dependent manner and are selectively downregulated by RNase P/MRP[26]. In innate immunity, Chen et al[27] found that unmodified circRNA adjuvant induces antigen-specific T and B cell responses, but m6A modification could abrogate circRNA immunity though YTHDF2 -mediated suppression. In male germ cells, the back splicing tends to occur mainly at m6Aenriched sites, which are usually located around the start and stop codons in linear mRNAs, resulting in about half of circRNAs containing large open reading frames.This potential mechanism could ensure long-lasting and stable protein production for specific physiological processes when lacking the corresponding linear mRNAs[28].These findings showed the important roles of m6A in circRNAs during disease progress. Therefore, it is essential to explore the roles of m6A circRNAs in SAP.
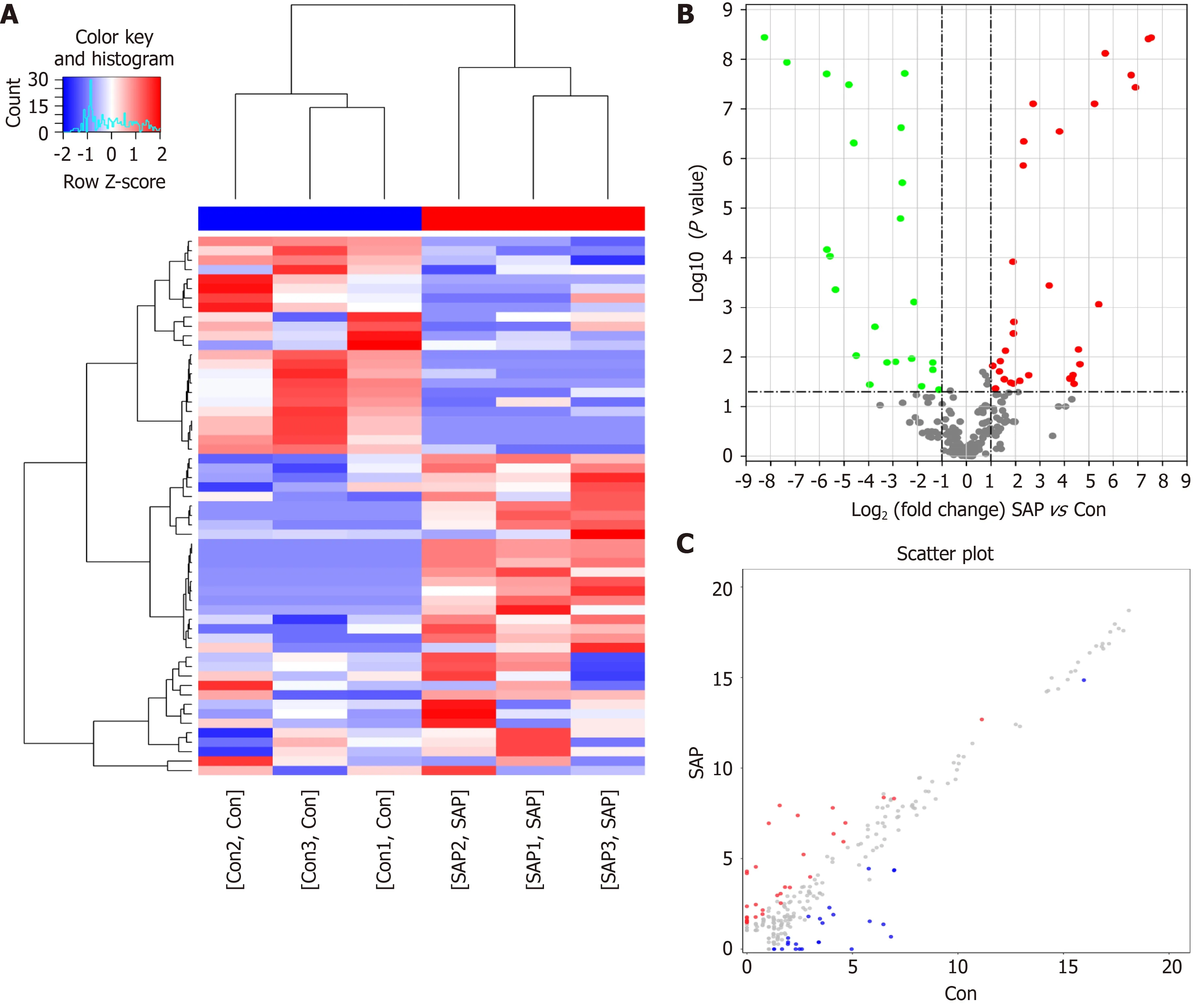
Figure 3 Differential N6 -methyladenosine modification of circRNAs in severe acute pancreatitis. A: Hierarchical clustering graph exhibiting differential N6 -methyladenosine (m6 A) modification of circRNAs in control and severe acute pancreatitis (SAP) groups. Higher expression is presented in red and lower expression in blue; B and C: Volcano and scatter plot showing the circRNAs with significant differentially expressed m6 A peaks.
In the present study, the function analysis of DE m6A circRNAs in SAP found that two important pathways were involved in the pathogenesis of SAP, including protein digestion and regulation of autophagy. As an important pathological cellular event,the activation of premature trypsinogen can result in acinar cell necrosis[1]. Many pancreatic injury factors, such as trauma, obstruction of the pancreatic duct and alcohol, can initiate the fusion of lysosomes with zymogen in acinar cells, leading to the activation of trypsinogen through cathepsin B to trypsin. Once trypsin is released,it can cause self-digestion in and outside the acinar cells, and the release of cathepsin B can cause necroptosis. As a cytoprotective mechanism, autophagy can process and recycle various aged, defective or damaged cytoplasmic contents[29]. Selective macroautophagy is a biological process during which specific damaged organelles and misfolded proteins are processed and recycled. Autophagy is accomplishedviaa series of steps, which start with the enucleation of cytoplasmic inclusions in the open double membrane formed by the ER, Golgi apparatus and plasma membrane[30]. Knocking outATG7genes (which are important to form autophagosome) andLAMPgenes could lead to pancreatitis with extensive inflammation in mice[29 ,31]. Importantly, impaired autophagy leads to trypsinogen activation, ER stress and mitochondrial dysfunction.These events can together make acinar cells become more susceptible to other insults and cellular death[1]. In addition, RNA transport is enriched in GO terms of note, and Chenet al[16] found that m6 A modification can modulate the export of circNSUN2 to the cytoplasm, suggesting that m6A modification regulates transport of circRNAs in SAP. These results were consisted with the hypothesis that m6A modification of circRNAs participated in the progression of SAP.
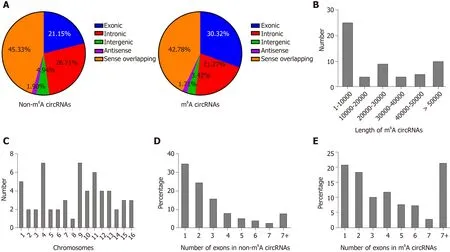
Figure 4 Distribution of N6 -methyladenosine sites in severe acute pancreatitis and control groups. A: Distribution of genomic origins of non-N6 -methyladenosine (m6 A) circRNAs (left) and m6 A circRNAs (right); B: Number of circRNAs with differentially expressed m6 A peaks based on the distribution of length;C: Chromosomal distribution of all differential m6 A sites within circRNAs; D and E: Distribution of non-m6 A and m6 A circRNAs based on the number of exons in each circRNA.
m6A modification of mRNA can influence its expression by regulating transcription,splicing and degradation[32]. In circRNAs, Zhou et al[14] and Su et al[33] reported that m6A levels are correlated with expression levels of circRNAs in HeLa cells and a rat model of hypoxia-mediated pulmonary hypertension. However, in SAP, we found m6A modification in circRNAs was not associated with expression of circRNAs,suggesting that m6A circRNAs function in SAP though other mechanisms, such as miRNA sponges. It is worth mentioning that more direct evidence is currently needed to support that m6A can affect circRNA expression.
miRNA sponges is an important function of circRNAs. Cytoplasmic circRNAs can prevent miRNAs from binding to target mRNAs by competitive binding to miRNA response elements, further playing a key role in diseases[8 ,34]. For instance, in lung squamous cell carcinoma, circTP63 can competitively bind to miR-873 -3 p and prevent miR-873 -3 p from decreasing the level of FOXM1 . The FOXM1 can upregulate the expression of CENPA and CENPB, ultimately facilitating cell cycle progression[35]. In SAP, circHIPK3 can enhance pyroptosis via regulating the miR-193 a-5 p/GSDMD axis in acinar cells, ultimately aggravating this disease[36]. In our previous study, we found that circZFP644 could sponge miR-21 -3 p, thereby participating in the pathogenesis of SAP[9]. Recently, Su et al[33] found that m6 A modification of circRNAs could influence the interactions between circRNAs and miRNAs. Therefore, analysis of m6A circRNA-miRNA networks was performed in this study. Several important miRNAs participated in the pathological process of SAP were found to bind to these m6A circRNAs, such as miR-24 -3 p, miR-26 a, miR-92 b, miR-216 b, miR-324 -5 p and miR-762 . For example, in caerulein-stimulated AR42 J cells, expression of miR-92 b-3 p was decreased, while overexpression of miR-92 b-3 p could downregulate the expression of TRAF3 and inhibit the MKK3 -p38 pathway, attenuating inflammatory response and autophagy[37]. These results suggest that m6 A modification of circRNAs functions by influencing the interactions between circRNAs and miRNAs.
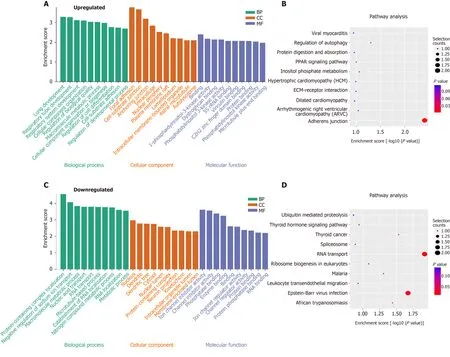
Figure 5 Functional analysis of circRNAs with differentially expressed N6 -methyladenosine peaks though gene ontology and Kyoto Encyclopedia of Genes and Genomes analysis. A and B: Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of circRNAs with upregulated N6 -methyladenosine (m6 A) peaks; C and D: GO and KEGG analysis of circRNAs with downregulated m6 A peaks. GO analysis include biological process (BP) analysis, cellular component (CC) analysis, and molecular function (MF) analysis.
m6A modification is a reversible process that occurs by methyltransferase complex consisting of METTL3 , METTL14 and WTAP, and is “erased” by ALKBH5 and FTO[13 ,15]. In pancreatic cancer, ALKBH5 could regulate the post-transcriptional activation of PER1 through m6 A abolishment, thereby inhibiting the cancer[38]. In hepatocellular carcinoma, ALKBH5 could attenuate expression of LYPD1 by an m6 Adependent manner and act as a tumor suppressor[39]. Overall, this evidence has suggested that ALKBH5 plays an essential role in m6 A modification. In this study, we found that expression level of ALKBH5 was upregulated in SAP. Consistent with this result, total m6A level of circRNAs in SAP was reduced, indicating that ALKBH5 may play a role in the dynamic process of m6A in SAP.
However, there are still limitations in our study. Firstly, furtherin vivoandin vitroexperiments are needed to further explore the m6A circRNA-mediated precise regulatory mechanisms in SAP. Secondly, the conservation analysis of the m6A circRNAs showed that these circRNAs may have similar roles in human SAP.However, their clinical significance and the results should be investigated further in SAP patients. Additionally, the precise mechanism of ALKBH5 in m6 A circRNAs during SAP needs to be studied. Actually, these are in our next plans to explore the roles of m6A circRNAs in SAP.
CONCLUSION
In conclusion, our study identified the transcriptome-wide profiling of m6A circRNAs in SAP and predicted their biological significance and possible potential mechanisms,providing new insights to explore the possible pathophysiological mechanism of SAP and seek new potential therapeutic targets.
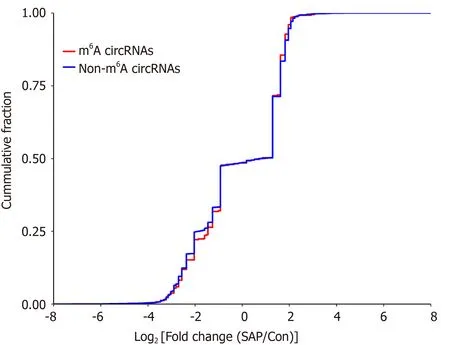
Figure 6 Relationship between N6 -methyladenosine level and expression of circRNAs in severe acute pancreatitis. Cumulative distribution of circRNAs expression between control and severe acute pancreatitis (SAP) groups for N6 -methyladenosine (m6 A) circRNAs (red) and non-m6 A circRNAs (blue).
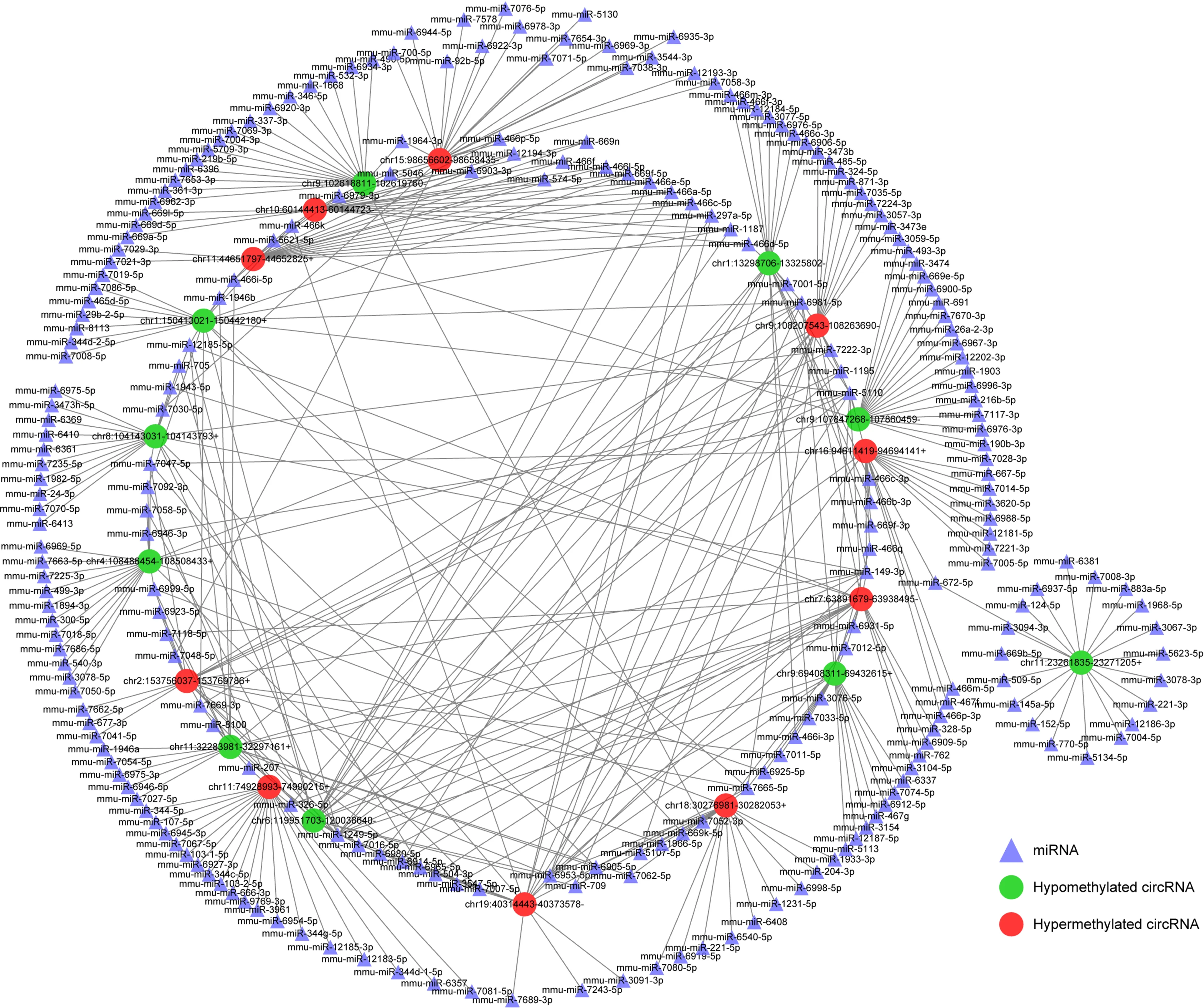
Figure 7 Construction of N6 -methyladenosine circRNA-miRNA networks in severe acute pancreatitis. A map showing the interaction networks of the top 10 upregulated and top 10 downregulated circRNAs according to the level of N6 -methyladenosine, and their around 20 target miRNAs with the most stable binding in SAP. Green circles represent hypomethylated circRNAs, red circles represent hypermethylated circRNAs and triangles represent miRNAs, compared with control group.
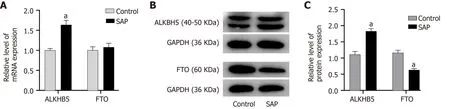
Figure 8 Expression of demethyltransferase in severe acute pancreatitis. A: Relative mRNA levels of alkylation repair homolog 5 (ALKBH5 ) and fat mass and obesity related protein (FTO) (normalized by the quantity of GAPDH) in each group; B: Representative images of western blot detected with alkylation repair homolog 5 (ALKBH5 ), FTO, and GAPDH antibodies in control and severe acute pancreatitis (SAP) groups; C: Relative protein levels of ALKBH5 and FTO(measured as the ratio of ALKBH5 , FTO to GAPDH by band density) in each group. Data are representative of at least three independent experiments. aP < 0 .05 vs control group.
ARTICLE HIGHLIGHTS


Research perspectives
This present study for the first time identified transcriptome-wide map of m6A circRNAs and determined their biological significance and potential mechanisms.However, the m6A circRNA-mediated precise regulatory mechanisms are need to be explore furtherin vivoand vitro experiments. What’s more, further studies are needed to reveal the precise mechanism of ALKBH5 in m6 A circRNAs during SAP. In the future, we will explore them and investigate these m6A circRNAs in SAP patients.
 World Journal of Gastroenterology2021年43期
World Journal of Gastroenterology2021年43期
- World Journal of Gastroenterology的其它文章
- Survivin-positive circulating tumor cells as a marker for metastasis of hepatocellular carcinoma
- Minimum sample size estimates for trials in inflammatory bowel disease: A systematic review of a support resource
- Immunoglobulin G in non-alcoholic steatohepatitis predicts clinical outcome: A prospective multi-centre cohort study
- Liver injury changes the biological characters of serum small extracellular vesicles and reprograms hepatic macrophages in mice
- Hepatitis B: Who should be treated?-managing patients with chronic hepatitis B during the immunetolerant and immunoactive phases
- Recent advances in artificial intelligence for pancreatic ductal adenocarcinoma