
头颈部副神经节瘤的遗传学研究进展
2021-12-01 07:19王璞,夏寅
协和医学杂志 2021年6期
王 璞,夏 寅
首都医科大学附属北京天坛医院耳鼻咽喉头颈外科,北京 100070
副神经节瘤是一种起源于外胚层神经嵴细胞的神经源性肿瘤,可发生于沿颅底至盆腔和骶椎的自主神经的任何部位,位于头颈部的副神经节瘤称为头颈部副神经节瘤(head and neck paraganglioma,HNPGL)。
HNPGL虽多无内分泌活性,但其血供丰富,且有多发性肿瘤倾向,手术具有一定难度[1];另有研究报道,可产生儿茶酚胺的HNPGL不足1%[2],应引起临床重视。HNPGL根据发病位置可进一步细分为颈动脉体瘤、颈静脉球瘤、鼓室体瘤和迷走神经副神经节瘤等[3-4]。HNPGL可发生于任何年龄段,30~40岁较常见,以女性患者居多,男女比例为1∶(3~4)。其发病率为1/10万~1/3万[5],约占头颈部肿瘤的3%[6],生长缓慢,多为良性,恶性率约为2%~9%,其中10%~15%可转移至肺、骨、肝和淋巴结等部位[5,7]。
副神经节瘤家系和散发病例均有报道,家系病例约占30%[8-9],提示遗传因素在副神经节瘤的发病中起一定作用。到目前为止,发现与HNPGL相关的基因共有10个(SDHA、SDHB、SDHC、SDHD、SDHAF2、VHL、HIF2A、RET、NF1和TMEM127),且不同基因的临床表型并不完全相同[3,10-12],比如遗传方式、外显率、发病年龄、肿瘤多发性及恶性率均不相同。了解副神经节瘤的分子机制及不同基因型-表型的相关性,有助于临床医生对HNPGL患者及家属进行遗传咨询、指导术前评估并制订治疗策略[13-14]。
1 分子机制
肿瘤基因组图谱将副神经节瘤按致病基因类型分为3类,即假性缺氧簇、Wnt信号簇和激酶信号簇[7,15]。
假性缺氧簇可分为两个亚组,第一亚组是与三羧酸 (tricarboxylic acid,TCA)循环相关的因子,约占嗜铬细胞瘤和副神经节瘤(pheochromocytoma and paraganglioma,PPGL)的10%~15%,包括编码琥珀酸脱氢酶4个亚基的基因SDHA、SDHB、SDHC、SDHD和一个稳定此4个亚基的基因SDHAF2,以及FH(TCA循环中的第二种酶)的胚系突变;第二亚组是与VHL/EPAS1相关的基因,占PPGL的15%~20%。假性缺氧组的特征是HIF的激活(对细胞缺氧的生理反应),由VHL、SDHx、EGLN1和HIF2A基因突变引起,其结果是尽管患者体内氧气水平正常,细胞仍处于缺氧状态。这一通路的激活将导致HIF靶基因的表观遗传学改变,包括细胞增殖、血管生成、迁移、凋亡和侵袭,最终导致PPGL的发生[15-16]。
Wnt信号簇主要由CSDE1基因的体细胞突变或影响MAML3基因的体细胞基因融合触发,这导致了Wnt和Hedgehog信号通路的激活。散发性PPGL (约占PPGL的5%~10%)多为这一组的基因异常。许多发育过程如增殖、细胞极性、黏附或分化,均受Wnt信号通路的调控。因此,此类肿瘤更具侵袭性,复发率高,且易转移[7,17-18]。
激酶信号簇是包括RET、NF1、MAX、HRAS和TMEM127基因的胚系或体细胞突变,约占PPGL的50%~60%。RET原癌基因激活或NF1抑癌基因失活可激活RAS/MAPK和PI3/AKT信号通路,导致肿瘤形成,而TMEM127基因突变激活了mTOR通路。另一种机制是MAX抑癌基因失活,从而使辅因子MYC(原癌基因)表达升高,最终导致PPGL的形成[16-18]。
2 基因型-表型相关性
目前,副神经节瘤的遗传因素共报道了17个易感基因,包括SDHA、SDHB、SDHC、SDHD、SDHAF2、VHL、HIF2A、RET、NF1、TMEM127、MAX、FH、KIF1B、EGLN1/PHD2、IDH1、HRAS和ATRX,其中前10个基因与HNPGL相关[15,19]。在10个HNPGL相关基因中,最常见的是SDHD和SDHB,其次是SDHC、SDHAF2和VHL,相对少见的是SDHA(报道2例)、RET(报道3例),比较罕见的是HIF2A、NF1和TMEM127,目前均仅报道1例。以下重点介绍10个基因的特点及其基因-表型相关性。
2.1 SDHD 突变倾向于形成多发性副神经节瘤
SDHD突变导致副神经节瘤1型综合征(paragangliomas syndrome 1,PGL1)的研究,可追溯至2000年,突变位于11q23.18[20]。Pasini等[21]2009年的荟萃分析显示,395例SDHD基因突变副神经节瘤患者中,多发性副神经节瘤较常见,成为该基因突变的一个典型特征,约占60%~79%;SDHD突变发生HNPGL的概率为91%~98%,发生交感性副神经节瘤的风险为16%~60%。SDHD突变在HNPGL较常见,大部分无内分泌活性,但仍有约20%会分泌多巴胺或其代谢产物[22],应引起重视。
SDHD突变的遗传方式为常染色体显性遗传,但带有明显的母亲印记,即从母亲遗传的突变不会发病,而从父亲遗传的突变很可能会发病;由于母亲有50%的概率遗传给下一代,因此会出现隔代遗传现象。母亲印记也在副神经节瘤2型(SDHAF2,11q13)中发生过,但未见于3型和4型,这可能与11号染色体有关[3,23]。
SDHD突变患者平均发病年龄为36岁。随着年龄的增长,SDHD突变发病率也在增长。2004年对SDHD突变的患者进行调查发现,31岁外显率为50%,50岁外显率升至86%[24]。2009年Hensen等[25]报道,SDHD突变在40岁时的外显率为54%,60岁时外显率为68%,70岁时外显率为87%。2012年,一项Meta分析发现,SDHD突变患者的恶性率为8%[26]。
2.2 SDHB 突变有交感性副神经节瘤及恶变倾向
SDHB突变与PGL4发病相关,位于1p36.13,为常染色体显性遗传[27]。SDHB突变多表现为交感性副神经节瘤(52%~84%),发生嗜铬细胞瘤的概率为18%~28%[28],发生HNPGL的概率为27%~31%[29]。SDHB突变发生多发性副神经节瘤的可能性不高,仅约为8%[29-30]。平均发病年龄39岁,外显率也与年龄相关,30岁时外显率为29%~30%,40岁时外显率为45%[22]。SDHB突变的患者表现为恶性嗜铬细胞瘤和恶性HNPGL的发生率为20.6%~41%[22-24,31]。2012年,van Hulsteijn等[26]通过系统性综述和Meta分析发现,SDHB突变的恶性率为17%。在多因素分析中发现SDHB是致死的唯一危险因素,有SDHB突变的患者5年生存率为36%,而无SDHB突变的5年生存率为67%[31]。
2.3 SDHC突变几乎100%表现为HNPGL
SDHC突变可导致PGL3,位于1q21,为常染色体显性遗传[28,32]。SDHC突变几乎100%表现为HNPGL,但有多发性副神经节瘤倾向,约占19%~31%[29,33-35]。SDHC突变发生率低,据两项大样本研究(n=598和n=445)报道,SDHC突变在HNPGL患者中的检测率分别为3.6%和4.3%[29,33],患者很少发生嗜铬细胞瘤或恶性肿瘤。SHDC突变患者中有家族史者约为12%~25%,说明外显率很低[33];平均发病年龄为38~46岁。
2.4 SDHAF2 突变倾向于形成多发性副神经节瘤
SDHAF2突变与PGL2相关,研究最早发表于2009年[36],其为常染色体显性遗传,有明显的母亲印记,突变位于11q13.1[30]。目前该基因突变仅见于HNPGL[37]。对含SDHAF2突变的荷兰大家系的30年随访研究发现,该家系3代72个家庭成员中,57例患者含SDHAF2基因突变,且发病年龄较早(平均发病年龄33岁),50岁时100%发病,均为HNPGL,其中91%有多发性副神经节瘤表现,未发现恶变病例[37-38]。
2.5 VHL 基因新发突变较多且部分为体细胞突变
VHL突变可引起VHL(von Hippel-Lindau)综合征,位于3p25.3。其是肿瘤抑癌基因,主要调控HIF的活性和细胞增殖,包括血管生成[28]。VHL综合征是一种常染色体显性遗传的多系统肿瘤综合征,该病平均发病年龄为26岁,65岁时外显率超过90%,平均生存年龄不足50岁,中枢神经系统血管母细胞瘤和肾细胞癌并发症为VHL患者最主要的死因[39]。VHL综合征根据临床表现可分为无嗜铬细胞瘤(1型)和有嗜铬细胞瘤(2型)2种类型,其中VHL2型又分为3个亚型,2A型为有嗜铬细胞瘤和中枢神经系统血管母细胞瘤,不伴有肾细胞癌;2B型伴有肾细胞癌;2C型仅有嗜铬细胞瘤,无其他疾病[40]。
HNPGL患者中VHL突变约为0.5%[41]。新发突变约为20%[42],在散发性PPGL中检测到体细胞VHL突变为14%[43]。
2.6 与HNPGL发病相关的5种少见基因
SDHA突变与PGL5相关,位于5p15.33,为常染色体显性遗传。在副神经节瘤患者中,SDHA突变率为4.5%[44],但SDHA引起HNPGL较少见[28]。
RET位于染色体10q11.2,是一种酪氨酸激酶受体,调节细胞增殖和凋亡,RET突变与PI3K/v-Akt信号和Ras/RAF/MAPK信号通路的激活相关[28,30],可导致多发性内分泌腺瘤病2型(multiple endocrine neoplasia 2,MEN2)的发生。MEN2患者通常首先被诊断为甲状腺髓样癌(medullary thyroid carcinoma,MTC),这也是最常见的临床表型。目前,仅在3例MEN2患者中发现有HNPGL,且HNPGL不会在MEN2患者中单独发生[43]。
HIF2A突变可引起Pacak-Zhuang综合征,位于2p21。2012年首次在2例患者中检测到HIF2A体细胞突变,1例为副神经节瘤,另1例为副神经节瘤和生长抑素瘤,2例患者均表现为红细胞增多症[45]。随后又发现了4例不同种族的该类型疾病患者,确认了Pacak-Zhuang综合征的存在。此类患者多在出生或儿童时即表现为红细胞增多症,并伴有多发性副神经节瘤和生长抑素瘤[46-47]。到目前为止,仅发现1例患者表现为HNPGL,为颈静脉球瘤[46]。HIF2A编码的缺氧诱导因子可调控能量代谢、铁代谢、红细胞生成、细胞发育、糖酵解和其他细胞功能。HIF2A突变影响脯氨酰羟基化,从而使HIF2A泛素化和降解失败。因此,HIF2A突变的蛋白质半衰期延长,导致下游靶向因子内皮素-1、促红细胞生成素、葡萄糖转运蛋白1或血管内皮生长因子上调,从而导致肿瘤发生[45]。
NF1基因位于染色体17q11.2,编码一种GTPase激活蛋白,通过激活Ras-GTPase而发挥作用,参与多种对细胞生长和分化至关重要的信号级联反应,其表达受损使Ras活性持续增加,导致细胞增殖。NF1突变可导致I型神经纤维瘤病(neurofibromatosis type 1,NF1)[48],以及MTC和类癌[42]。最新研究发现,体细胞NF1突变与散发性PPGL相关[49]。早在1987年,曾报道1例NF1综合征患者患HNPGL[50],但到目前为止,尚无大样本研究发现NF1患者具有HNPGL的表现[29]。
TMEM127是一种肿瘤抑制基因,位于染色体2q11.2。研究表明,在哺乳动物中敲低TMEM127会影响mTORC1的表达,导致mTORC1的靶点磷酸化增加[28]。TMEM127突变与嗜铬细胞瘤相关。对PPC患者的遗传学研究表明,TMEM127突变的患病率较低(约为2%),在大多数情况下,TMEM127突变患者仅表现为嗜铬细胞瘤 (单侧和双侧),并分泌高水平的肾上腺素,仅1例表现为双侧HNPGL的病例报道[41,51-52]。
3 小结与展望
近年来对副神经节瘤的遗传学研究表明,5个基因突变(SDHB、SDHC、SDHD、SDHAF2和VHL)在HNPGL中较常见(表1)。其中,SDHD和SDHAF2突变易导致多发性肿瘤,提示临床医生应注意患者整个头颈部和全身情况,仔细检查有无多发性副神经瘤的体征和胸腹部隐匿肿瘤所引起的不典型症状;SDHB基因突变易导致交感性副神经节瘤且有恶变倾向,是致死的唯一危险因素,提示对此类患者应重点关注肿瘤转移情况并尽早手术,术后需密切随访;SDHD、SDHC和SDHAF2基因突变几乎100%表现为HNPGL,但SDHC基因突变在HNPGL中检出率较低,为3.6%~4.3%,而SDHAF2突变目前尚无大样本的报道;此外,SDHD和SDHB突变的外显率与年龄相关,年龄越大,外显率越高。目前,国内尚缺乏大样本量HNPGL患者基因检测的报道,未来需加强对HNPGL患者的基因检测,总结国内人群基因型与表型之间的关系,为HNPGL患者提供可靠的遗传学咨询,并进一步指导临床诊疗。
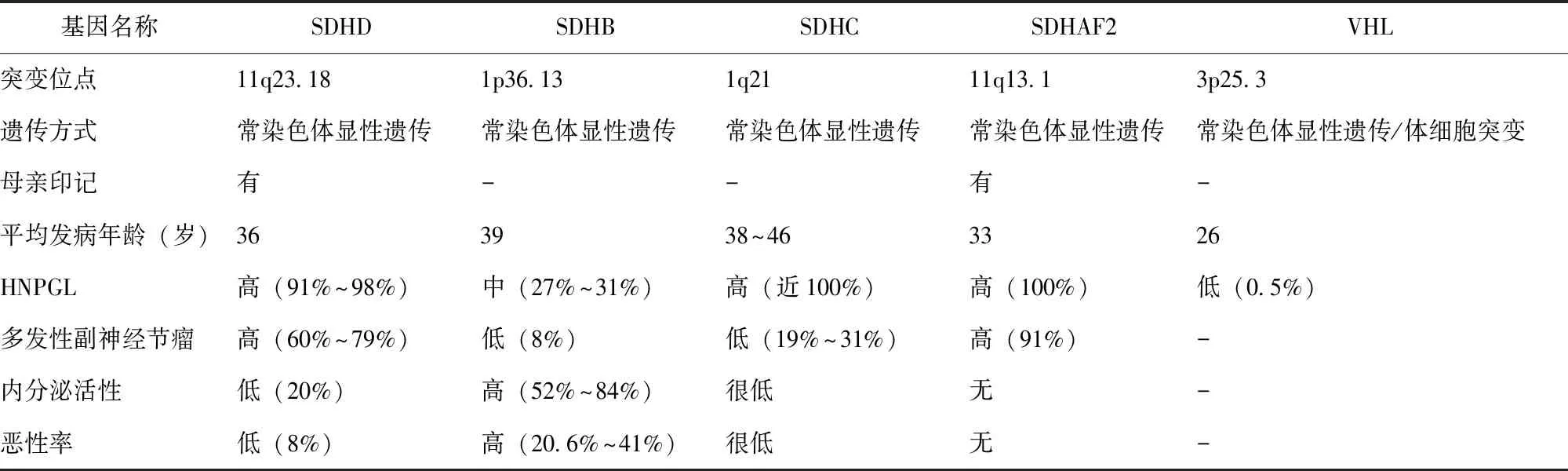
表1 与HNPGL发病相关的5种常见基因及其特点
作者贡献:王璞负责论文构思及撰写;夏寅指导论文修订。
利益冲突:无
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
中华养生保健(2020年7期)2020-11-16
临床肝胆病杂志(2020年9期)2020-09-28
中国临床医学影像杂志(2019年5期)2019-08-27
中医眼耳鼻喉杂志(2019年2期)2019-04-13
现代泌尿生殖肿瘤杂志(2018年5期)2018-11-29
健康管理(2016年6期)2016-05-14
中国医学影像学杂志(2015年9期)2015-12-15
中国医疗美容(2015年1期)2015-07-12
中国卫生标准管理(2015年17期)2015-01-26
