
高比表面积阿魏酸多孔淀粉酯结构表征及体外消化特性
2021-11-29 11:24苟丽娜马云翔王宇霞李敏陈金凤汪月张盛贵
食品与发酵工业 2021年22期
苟丽娜,马云翔,王宇霞,李敏,陈金凤,汪月,张盛贵
(甘肃农业大学 食品科学与工程学院,甘肃 兰州,730070)
阿魏酸(ferulic acid,FA)作为分布最广泛的天然酚类植物化学物质,多与木质素及多糖共价连接构成种子、叶片的部分细胞壁,普遍存在于蔬菜、水果、中草药及谷物种子中[1-2],由于其具有抗炎、抗病毒、抗过敏、抗癌、神经保护和抗血栓等[3-4]多种生物和药理活性,在食品及医药领域内引起了极大的关注。基于化学结构中苯环上羟基以及供电子体甲氧基的邻位取代,从而具备苯氧自由基稳定性[5],FA被证实是一种有效的抗氧化剂,其抗氧化活性包括清除自由基以及防止自由基链反应和脂质过氧化[6]。然而FA化学结构中苯环和短不饱和烃链的存在导致了低极性和低水溶性,使其难以渗透脂质双分子层,限制了其体内抗氧化作用[4]。但是,通过与淀粉、壳聚糖等生物可降解材料发生酯化或醚化反应进行化学修饰,可增强FA的水溶性和生物利用度[7-8]。
淀粉具备成本低廉、安全性、生物相容性、可降解性及化学惰性等优点,可作为理想的活性物质递送载体。但是,天然淀粉(native starch,NS)颗粒有限的比表面积和孔体积,限制了其在吸附和转运目标化合物方面的应用潜力。多孔淀粉(porous starch,PS)是一种通过化学、物理或酶处理后,淀粉颗粒表面及内部形成纳米级多孔微结构的改性淀粉。多孔结构提供的大的比表面积、高孔体积和低密度使PS具备强吸附性。在食品、医药工业中,PS已被用于不同活性物质的递送制剂,如改善疏水性活性剂溶解性、靶向药物递送、疏水性药物的吸附等[9]。SOLEIMANPOUR等[10]研究发现将水溶性差的植物雌激素染料木黄酮装载到PS中,可显著提升植物雌激素的溶解性、稳定性和生物利用度,其高吸附容量显示了多孔淀粉作为活性剂载体的潜在优势。
通过酯化、醚化、交联和接枝等衍生化反应将特定的官能团引入淀粉分子是最常见的淀粉化学改性方法[11-12],其中淀粉酯是食品和非食品应用中最重要的淀粉衍生物之一。NS特殊的半结晶结构,使得试剂难以渗透到颗粒中,导致反应效率较低[13]。相比NS,以PS作为反应主体,试剂分子渗透入孔隙、孔道和空腔中,淀粉内部暴露出更多的反应位点与客体分子发生反应,提高反应效率,进而获得更高的取代度。本团队先前研究表明,相同条件下辛烯基琥珀酸酐酯化改性多孔淀粉较天然淀粉的取代度更高[14]。
尽管已有阿魏酸酯化改性淀粉的研究但尚未见阿魏酸对多孔淀粉酯化改性的报道。本研究以天然马铃薯淀粉为原材料,制备纳米级孔径的PS,在有机体系中使FA与PS酯化,构建高取代度且具备大量孔隙结构的阿魏酸多孔淀粉酯(ferulic acid porous starch ester,FA-PS),对FA-PS的结构性能进行表征,并研究其体外消化特性,旨在为淀粉的综合利用提供理论依据,且为改性淀粉作为潜在的活性物质吸附剂和功能性递送载体材料提供一个有吸引力的选择。
1 材料与方法
1.1 材料
天然马铃薯淀粉、阿魏酸、二甲基亚砜(dimethyl sulfoxide, DMSO)、氯化亚砜、吡啶、无水乙醇、2,2-二苯基-1-苦肼基(DPPH)、胰酶、淀粉葡糖苷酶(AMG,100 000 U/mL)及其他试剂均为分析纯,上海麦克林生化科技有限公司;DMSO-d6(纯度≥99.9%),由剑桥同位素实验室提供。
1.2 仪器与设备
HCJ-4D型恒温磁力搅拌水浴锅,常州市瑞华仪器制造有限公司;ASAP 2020型全自动比表面积及孔隙度分析仪,美国麦克公司;NEXUS-670型傅立叶红外光谱仪,美国Thermo公司;UV-2550型紫外可见分光光度计,日本Shimadzu公司;JSM-6701F型冷场发射型扫描电子显微镜,日本电子光学公司;X′Pert-Pro MPD型多晶粉末X射线衍射仪,荷兰PANalytical公司;AVANCE III HD 400 MHz型超导核磁共振波谱仪,瑞士Bruker公司。
1.3 实验方法
1.3.1 马铃薯多孔淀粉的制备
参考CHANG等[15]的方法并改进,称取15 g马铃薯淀粉加入100 mL蒸馏水中,40 ℃磁力搅拌40 min 使其均匀分散,升温至90 ℃并持续搅拌60 min 使淀粉完全糊化,自然冷却至室温后置4 ℃冷藏48 h以获得淀粉凝胶,将淀粉凝胶切成立方体小块(0.6~0.8 cm3),在室温下经5次乙醇置换后热风干燥(50 ℃,6 h),研磨备用。
1.3.2 FA-PS的制备
参考李颜利[16]、MATHEW等[17]的方法并改进,采用一步法合成FA-PS,具体操作如下:
称取一定量FA至100 mL三口烧瓶,加入40 mL二氯甲烷磁力搅拌使其分散均匀,将与FA等摩尔量的氯化亚砜稀释在10 mL二氯甲烷中,在N2保护下逐滴滴入三口瓶中,之后升温至80 ℃回流反应4 h,停止反应。反复蒸馏除去过量的氯化亚砜和溶剂,得到阿魏酰氯。
将一定量PS分散在DMSO中,加入得到的阿魏酰氯中,以相对酰氯2∶1的摩尔比加入吡啶,在N2保护下80 ℃磁力搅拌状态连续回流反应3 h,待反应液冷却至室温,边搅拌边缓慢加入无水乙醇(300 mL×3)析出沉淀物,离心除去上清液,收集固体,用无水乙醇索氏抽提36 h,真空冷冻干燥48 h,研磨后过100目筛备用。
1.3.3 取代度(degree of substitution,DS)测定
DS的测定和计算参考NAMAZI等的方法[18]。称取一定量样品于核磁管中,加入DMSO-d6,并超声30 s使溶解充分并去除溶解氧对实验的影响,使用核磁共振波谱仪TXISz探针扫描测定,温度25 ℃。
在1H核磁共振图谱中,5.11处的峰为脱水葡萄糖单元(anhydro glucose unit,AGU)中的质子(H1)信号,阿魏酰基链上甲氧基(—OCH3)端的质子信号峰在3.83处。由3.83处的质子峰面积与5.11处质子峰面积关系,可通过公式(1)计算样品DS:
DS=(Asignal/Nsignal)/(AAGU/NAGU)
(1)
式中:Asignal、Nsignal,分别是样品在3.83处的峰面积、质子数;AAGU、NAGU,分别是样品在5.11处的峰面积、质子数。
1.3.4 N2-吸附/脱附等温线测定
利用Micromeritics ASAP 2020系统测定样品的N2-吸附/脱附等温线。测量前先将样品置于120 ℃下真空脱气8 h,除去样品表面吸附的气体分子和残余的溶剂。利用非定域密度泛函理论,从吸附数据中得到孔径的尺寸分布曲线。样品的比表面积依据Brunauer-Emmett-Teller(BET)多层吸附理论计算。
1.3.5 扫描电子显微镜(scanning electron microscope,SEM)表观形态观察
用无水乙醇将少量样品粉末分散在样品座上,将样品座置于离子溅射仪中镀金60 s后,用扫描电子显微镜在不同放大倍数下观察样品的形态特征。
1.3.613C交叉极化/魔角旋转固体核磁共振(13C cross polarization/magic angle spinning nuclear magnetic resonance,13C CP/MAS NMR)光谱测定
使用400 MHz超导核磁共振波谱仪,在发射机频率为100.56 MHz下以四甲基硅烷的13C化学位移为参照测定样品粉末的13C CP/MAS NMR光谱。
1.3.7 傅立叶变换红外光谱(Fourier transform infrared spectroscopy,FT-IR)测定
样品的红外光谱测定采用溴化钾压片法。取适量溴化钾加入1 mg干燥至恒重的样品粉末混合均匀,碾磨至均匀无颗粒感后压片处理,在4 000~500 cm-1波数范围内进行扫描测定。
1.3.8 X射线衍射(X-ray diffraction,XRD)光谱测定
采用X射线衍射仪对样品粉末在5~35°范围内进行连续扫描测定。
1.3.9 自由基清除活性测定
参照BRAND-WILLIAMS等[19]的方法稍做修改,使用DPPH测定样品粉末的自由基清除活性。分别称取1、3、5、7、9、11、13 mg(干基)样品粉末于10 mL试管中,加入2×10-4mol/L的DPPH乙醇溶液4 mL,漩涡振荡2 min,在室温黑暗条件下静置30 min后离心10 min(7 000 r/min)取上清液,以乙醇为空白对照,在517 nm处测定吸光值。DPPH自由基清除率可通过公式(2)计算:
(2)
式中:A0,为空白样吸光值;A1,样品吸光值;A2,乙醇溶液代替DPPH溶液所测吸光值。
1.3.10 水溶性的测定
为检测样品水溶性,称取30 mg样品粉末通过温和搅拌将溶于5 mL蒸馏水中30 min,观察溶解情况,在4 ℃冰箱中贮存24 h后离心并观察样品在水中的不同溶解情况及是否有沉淀物。
1.3.11 体外消化率测定
样品的体外消化率测定参考LEE等[20]的方法稍做修改,称取1 g胰酶分散在12 mL蒸馏水中10 min后以1 500 r/min离心10 min,取10 mL上清液与0.2 mL淀粉葡糖苷酶和1.8 mL蒸馏水混合(酶液现配现用)。将一颗玻璃小球(直径5 mm)与30 mg样品加入试管,然后加入pH为5.2的乙酸钠缓冲液4.5 mL,在37 ℃恒温水浴锅中恒温10 min,然后将1.5 mL酶溶液加入样品管中,在37 ℃和150 r/min 振荡水浴中水解20和120 min时,分别取水解液1 mL加入离心管,沸水浴5 min灭酶,离心10 min(1 500 r/min)后取上清液,采用DNS比色法,测定产生的游离葡萄糖含量。样品中的淀粉含量为淀粉水解液中还原糖含量乘以葡萄糖折算系数0.9。各类淀粉含量可通过公式(3)、公式(4)、公式(5)计算:
(3)
(4)
抗性淀粉含量/%
=(1-快消化淀粉含量-慢消化淀粉含量)×100
(5)
式中:FG,原始样品中游离葡萄糖的含量,mg;G20、G120,分别是消化20、120 min后释放的葡萄糖的含量,mg;m,每次实验所用的样品质量,mg。
1.4 统计分析
每组实验均重复测定3次,采用SPSS 19及Origin 8进行数据统计分析处理,结果以平均值±标准偏差表示。
2 结果与分析
2.1 多孔淀粉的孔结构表征
2.1.1 多孔淀粉N2-吸附/脱附分析
为研究PS的孔结构特性,对N2-吸附/脱附等温线数据进行分析。国际纯粹与应用化学联合会根据孔径大小将孔隙分为三大类:微孔(<2 nm)、介孔(2~50 nm)、 大孔(>50 nm),根据国际纯粹与应用化学联合会分类,PS的N2-吸附/脱附等温线对应为IV型[21],如图1-b所示,表现出典型的毛细冷凝滞后回线,滞后环陡峭狭窄,出现在0.5~1.0 (P/P0)的相对高压范围内,这与毛细管冷凝机制对介孔结构孔的填充和排空有关。而图1-a中NS的N2-吸附/脱附等温线中并未观察到毛细冷凝滞后回线,表明PS具备介孔结构。

a-天然马铃薯淀粉; b-多孔淀粉
为进一步表征制备材料的孔径,采用Barret、Joyner和Halenda(BJH)方法计算孔隙分布,如图2-a所示,PS的平均孔径为20.23 nm,位于介孔材料孔径分布(2~50 nm)范围内,验证制备的PS可归属于介孔材料范畴。图2-b、图2-c中利用Brunauer Emmett Teller(BET)模型计算出NS的比表面积仅为0.02 m2/g, 而PS的比表面积达到67.74 m2/g。
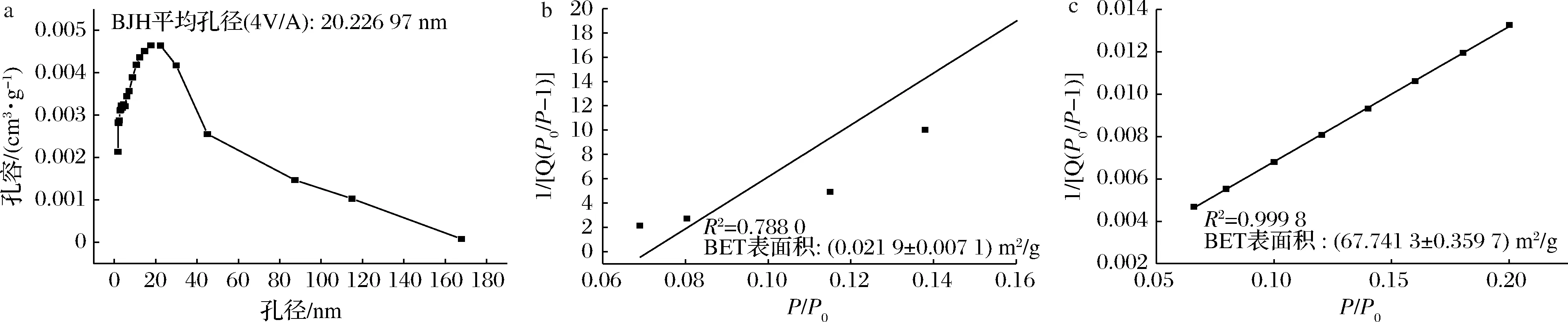
a-多孔淀粉孔径分布图; b-天然马铃薯淀粉BET表面积图; c-多孔淀粉BET表面积图
2.1.2 多孔淀粉SEM形态结构分析
为了分析淀粉改性前后表观形态的差异,使用SEM观察了淀粉样品的显微结构,如图3所示,成孔反应明显改变NS原有微观形貌。不同放大倍数下NS颗粒外形为表面光滑、比较规整且结构致密的球形(图3-a、图3-b),PS颗粒表面粗糙,规整度很差,与NS相比,其表面形成大量分布不均的海绵状或泡沫状孔隙结构(图3-c、图3-d),其成因是天然马铃薯淀粉糊化后冷藏形成包容着大量水分的水凝胶,在溶剂交换过程中,乙醇流入水凝胶中形成醇凝胶,蒸发时乙醇逃逸形成孔隙和泡沫结构,与水凝胶比较,醇凝胶在直接干燥过程中能避免孔隙的塌陷,有助于保持多孔状结构[22]。

a-天然马铃薯淀粉×5 000; b-天然马铃薯淀粉×30 000; c-多孔淀粉×5 000; d-多孔淀粉×30 000
2.2 阿魏酸多孔淀粉酯结构表征
2.2.1 阿魏酸多孔淀粉酯取代度(DS)
淀粉聚合物链中每个AGU中C2、C3、C6上3个游离羟基均可作为位点与羧酸发生酯化反应[12]。酯化反应中,吡啶作为碱性亲核酰化催化剂,DMSO的溶胀作用激活暴露的羟基,使PS受到亲电试剂的攻击。淀粉颗粒具备大的比表面积暴露更多化学反应活性位点,对客体分子的敏感性增大[23],进而提升反应效率获得更高的取代度。如表1所示,FA-PS的DS随FA与PS反应摩尔比增加而增大,反应中阿魏酰氯浓度越高,淀粉附近的分子碰撞率越高,分子的利用率就越高。化学改性淀粉的安全性一直是食品及医药应用中的首要考虑的问题,李颜利[16]的研究表明,在实验浓度范围内,先制备阿魏酰氯再酯化多糖得到的酯化物本身对多种细胞(人体肝癌细胞HepG2、人胰腺上皮细胞EC、成年人皮肤成纤维细胞HSF)无毒性。
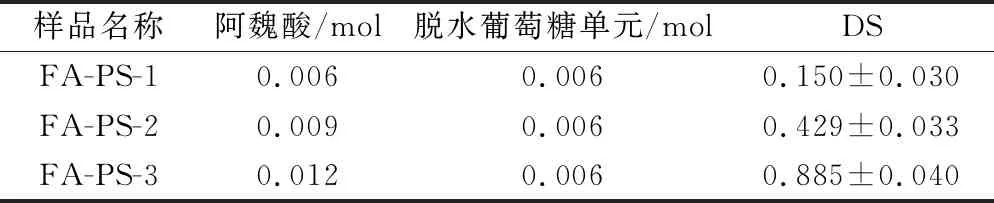
表1 阿魏酸多孔淀粉酯的DS
2.2.2 SEM形态结构分析
酯化改性后,淀粉颗粒表面粗糙,呈泡沫状多孔状结构。酯化反应在非水相体系中进行,可避免孔结构因水的溶胀作用及糊化作用而崩塌,图4-a中低DS的FA-PS孔大小不一,分布不均匀,而比较图4-b、图4-c,高DS淀粉酯的孔呈蜂窝状絮状并趋于均匀致密化分布,结果表明淀粉酯化反应强度与结构有较强的关联性,可能与酯化反应发生在淀粉表面及淀粉颗粒内部有关,酯化前后SEM结果显示样品表面形貌产生差异的原因需要进一步研究。

a-FA-PS-1×5 000; b-FA-PS-2×5 000; c-FA-PS-3×5 000
2.2.3 N2-吸附/脱附分析
相比于PS,图5-a中FA-PS-3的回滞环变宽而稍缓,毛细冷凝滞后现象较PS更强。FA-PS-3的平均孔径仅为8.59 nm,较PS平均孔径缩小了57.52%,属于介孔材料(图5-b),比表面积为83.50 m2/g,相较于PS提升了23.27%(图5-c)。结果表明酯化反应后形成了更小而均匀致密的孔,此结果与SEM观察到的结果一致,XU等[24]也观察到类似的结果,这可能是由于PS发生酯化反应使得淀粉羟基被取代。孔淀粉的负载量随比表面积的增加而增大,适当减小孔径可以增强毛细作用力有助于更高的负载[25]。

a-N2-吸附/脱附等温线; b-孔径分布图; c-根据等温线计算的BET表面积图
2.2.413C CP/MAS NMR分析
为了确认酯化反应过程中FA与PS之间酯键的形成,对PS和FA-PS-3进行13C CP/MAS NMR分析,根据文献给出了多孔淀粉分子中碳原子的对应信号[26]。如图6所示,酯化反应后,FA-PS-3的淀粉骨架结构保持良好。随着酯化反应的进行,淀粉上引入了阿魏酰基,与PS相比,FA-PS-3谱图中除了淀粉AGU的信号峰外,在109~160范围内的碳信号来自于阿魏酰基,165处出现羰基信号,这是由酯键的13C化学位移引起的,55处出现的峰为阿魏酰基上甲氧基的信号峰[17],结果表明,通过酯化反应成功形成了FA和PS之间的骨架连接。
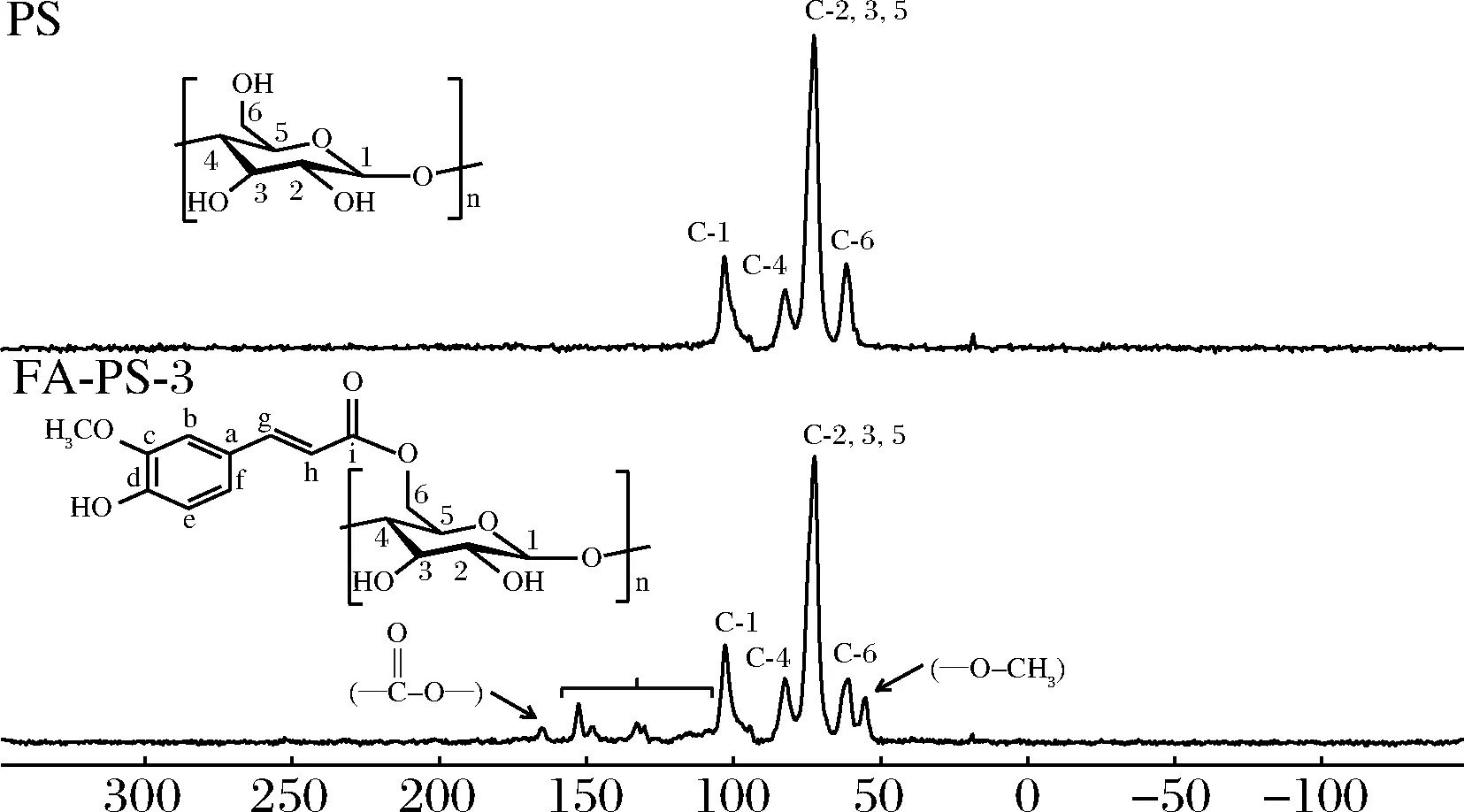
图6 多孔淀粉和阿魏酸多孔淀粉酯的13C CP/MAS NMR谱图
2.2.5 FT-IR分析

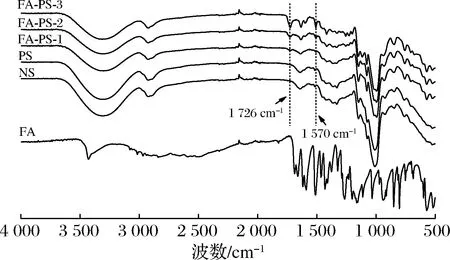
图7 阿魏酸、天然马铃薯淀粉、多孔淀粉及阿魏酸多孔淀粉酯的红外光谱图
2.2.6 XRD分析
改性反应对淀粉结晶结构的影响如图8所示,NS的X射线衍射图中5.2°、15.2°、17.2°、22.9°处存在较强的衍射峰,表明天然马铃薯淀粉属于B型结晶。而PS的图谱中仅观察到对应于无定形物质的分散性宽峰,淀粉高度有序的晶体结构由分子内和分子间的氢键负责,PS在制备过程中糊化作用使淀粉微晶熔融,导致有序的晶相转变为非晶无序状态,淀粉的稳定氢键断裂和晶体结构的破坏,可导致活性和游离羟基的产生,这有助于增加淀粉的反应性[28]。FA-PS 在13.0°、17.9°、20.4°、25.7°处观察到新的衍射峰,表明酯化作用形成新的晶体结构,且峰值强度随着DS的增强而增大。其晶型回生可能由于淀粉颗粒崩解形成的短链直链淀粉在回生过程中重新聚集并结晶,直链淀粉分子可形成双螺旋进而形成结晶域,具体形成机理有待进一步研究。
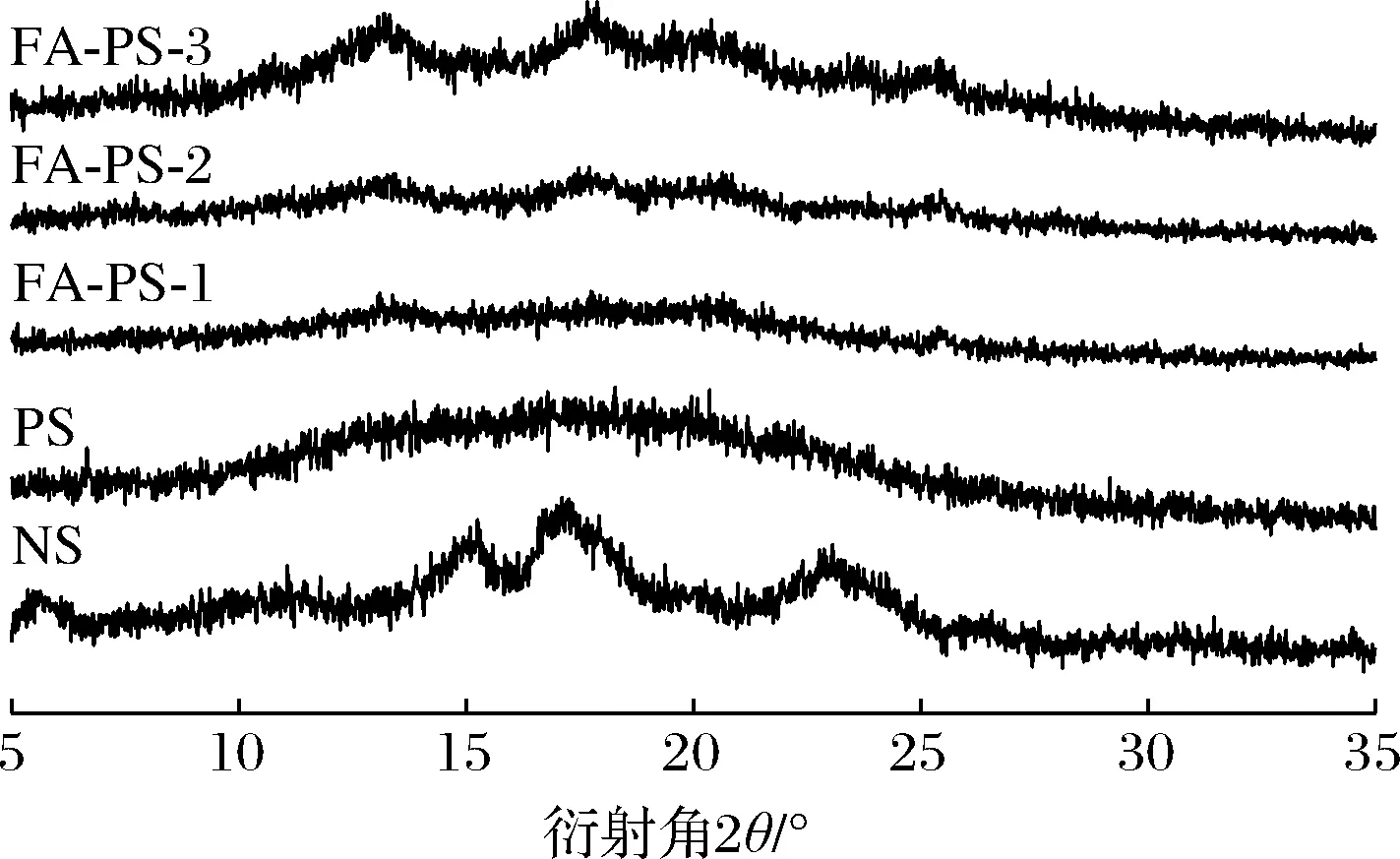
图8 天然马铃薯淀粉、多孔淀粉及阿魏酸多孔淀粉酯的X射线衍射图
2.3 DPPH自由基清除能力分析
为了评估酯化改性后FA-PS的抗氧化活性,测定PS和FA-PS对DPPH自由基的清除率如图9所示,PS几乎没有清除DPPH自由基的能力,而FA-PS对DPPH自由基的清除率随体系浓度和DS的增加而增强,FA-PS-3对DPPH自由基的清除率最高达到67.76%,WEN等[29]也观察到类似结果。酯化反应后淀粉聚合物链上接入的阿魏酰基是抗自由基活性的决定因素,由于酚羟基与自由基反应形成共振稳定的苯氧基,丙烯酸侧链的共轭双键可以通过共振对苯氧基产生稳定作用,供电子体甲氧基有效加强苯氧基稳定性[6],使FA-PS具备了较强的抗氧化活性。

图9 多孔淀粉和阿魏酸多孔淀粉酯的DPPH自由基清除能力
2.4 水溶性
淀粉改性后水溶性改善情况如图10所示,由于化学结构中苯环和短不饱和烃链的存在,阿魏酸在水性介质中溶解性很差[6]。天然马铃薯淀粉不溶于冷水,静置后沉淀于试管底部,多孔化改性后的高比表面积大大增加亲水性表面,PS的水溶性有所改善,酯化改性后FA-PS的水溶性得到了明显的提升,且随淀粉酯取代度增加,水溶性逐渐增强。对于大多数低溶解度和高胃肠道渗透性的活性剂,其体内生物利用度可通过提高溶解度有效提升[9]。该研究结果表明,FA-PS有望应用于食品和医药相关领域。

图10 天然马铃薯淀粉、多孔淀粉、阿魏酸及阿魏酸多孔淀粉酯的水溶性
2.5 体外消化性能分析
淀粉基载体摄入后会在口腔和肠道阶段被淀粉酶侵蚀,从而限制其通过口服途径递送活性物质的位点依赖性释放能力。为研究多孔化和酯化改性对淀粉体外消化性能的影响,测定NS、PS和FA-PS的体外消化率,结果如图11所示。与NS相比,PS的快消化淀粉和慢消化淀粉含量分别为21.68%和27.56%,均显著增高,抗性淀粉含量为50.75%,显著降低(P<0.05)。 PS中孔隙、孔道和空腔的存在增加了酶反应的表面,消化酶水解的敏感性增强,同时多孔化改性后PS的结晶结构处于不定形状态,通常分子无序、松散堆积可能容易结合淀粉酶进行水解,淀粉中这种不明确的分子排列顺序更容易被酶消化[30]。基于阿魏酰基的化学修饰,随DS增大,FA-PS的快消化淀粉和慢消化淀粉含量逐渐减少,抗性淀粉含量增加。FA-PS-3的抗性淀粉含量高于NS,显著高于PS(P<0.05),达到73.85%。FA-PS的高比表面积同样提供了更多酶反应位点,但是,研究表明FA可以抑制淀粉消化酶的催化效率[5-6]。同时PS与FA酯化时分子重排影响淀粉消化速率,淀粉酯中聚合物分子有序化排列减少了淀粉消化酶与活性位点的结合,有效阻止了酶对淀粉的水解,导致与PS相比,FA-PS的体外抗消化性能显著提升。
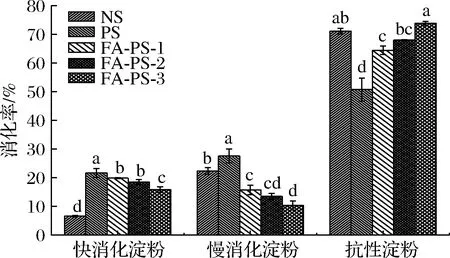
图11 天然马铃薯淀粉、多孔淀粉及阿魏酸多孔淀粉酯的体外消化率
3 结论
猜你喜欢
小学生学习指导(高年级)(2022年3期)2022-03-29
能源化工(2021年6期)2021-12-30
酿酒科技(2021年5期)2021-06-06
上海理工大学学报(2020年5期)2020-11-21
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
小学生学习指导(高年级)(2019年4期)2019-11-27
科学中国人(2018年8期)2018-07-23
小学生导刊(高年级)(2017年2期)2017-06-10
小学生导刊(高年级)(2016年1期)2016-01-29
