
天然产物中炔基的生物合成机制研究及其应用
2021-11-29 06:40吕建明赵欢胡丹高昊
合成生物学 2021年5期
吕建明,赵欢,胡丹,高昊
(1 暨南大学药学院,中药及天然药物研究所,广东 广州 510632; 2 暨南大学中医学院,广东 广州 510632)
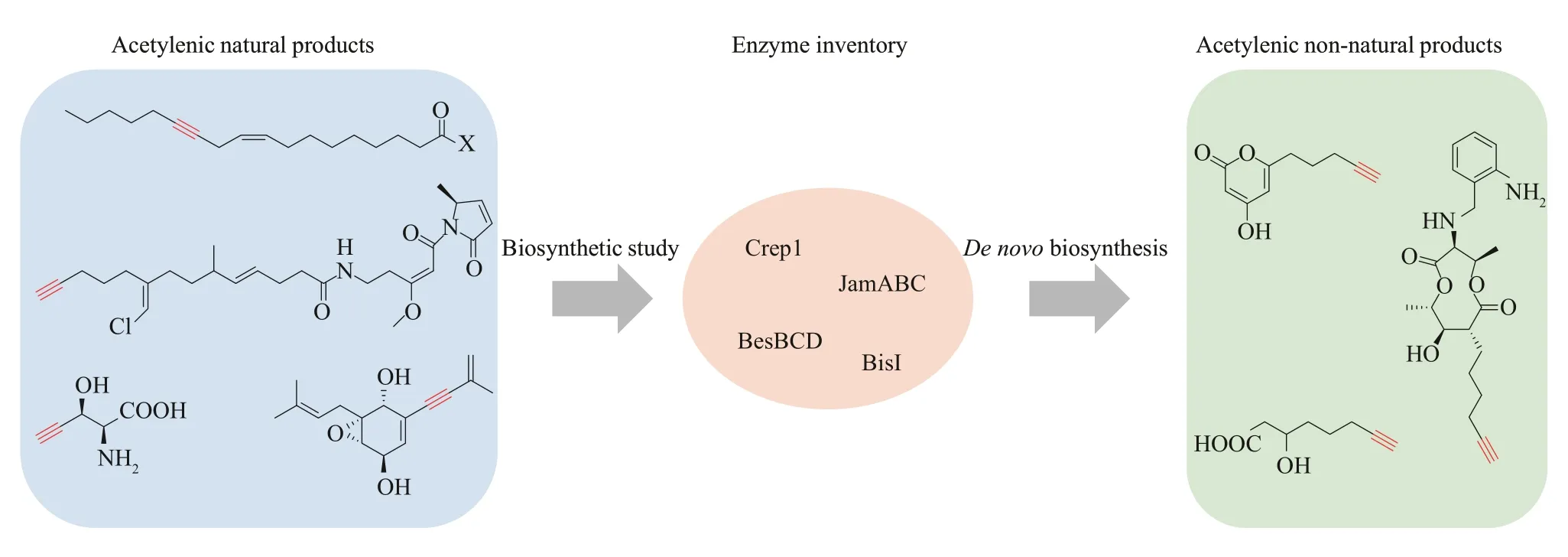
炔基是许多临床一线药物的重要功能基团[1],如 FDA 批准的第 1 个口服避孕药 Enovid®[2]以及用于治疗艾滋病毒感染的第1代非核苷类逆转录酶抑制剂Sustiva®[3]等的主要成分都是含有炔基的小分子化合物[4-7]。自然界中也存在大量具有广泛生物活性的炔类天然产物[8-11],如从箭毒蛙的皮肤中分离获得的histrionicotoxin 类生物碱是烟碱型乙酰胆碱受体的非竞争性抑制剂[12],从多种伞形科植物中分离获得的falcarinol 类聚炔化合物具有抗肿瘤、抗炎以及抗菌等活性[13-14],从细菌中分离获得的烯二炔类抗生素能够显著抑制肿瘤细胞生长[15-16]。此外,炔基还可参与一系列的化学反应[17-21],是重要的化学合成模块,广泛用于活性天然产物[22-23]以及功能材料[24]等的合成。特别是基于末端炔基与叠氮化物的环加成反应(即点击化学反应),在药物筛选、化学生物学以及蛋白组学等研究领域发挥着重要的作用[25]。
由于炔基是一个非常重要的结构单元,因此开发高效的炔类化合物合成策略具有重要的意义。目前最常用的策略是通过化学方法直接制备含有炔基的目标产物,但其不足之处是反应条件严苛、成本高以及反应效率低等[26-27]。另一策略是生物转化,即将炔类化合物作为底物添加到微生物培养体系中或在体外与酶共孵育,通过生化反应把炔基整合到目标化合物中[28-31]。但是由于炔类前体通常不易获得,该策略存在一定的局限性。合成生物学是指基于工程化设计思路,通过构建标准化的元器件和模块来改造天然系统或从头合成全新的人工生命体系,其能将结构简单、易于获得、廉价的起始原料高效地转化成不同的高附加值产品,而且操作简单、绿色环保,在化学品合成领域有极大的应用潜力[32-33]。因此,基于合成生物学技术的从头合成有望成为对传统化学合成以及生物转化策略的有效补充,但其成功实施的前提是获得催化天然产物中炔基形成的酶,即炔基合成酶。近年来,研究人员开展了不同炔类天然产物的生物合成研究,以期为炔类化合物的生物制造提供丰富的酶工具。本文综述了不同天然产物中炔基的生物合成研究现状,然后介绍了不同炔基合成酶在炔类化合物从头生物合成方面的应用情况。
1 天然产物中炔基的生物合成
人们从动物、植物与微生物中分离鉴定了大量不同类型的炔类天然产物[8,34-35],包括脂肪酸类、聚酮类、聚酮-非核糖体肽杂合体类、氨基酸类、杂萜类等,而且炔基在同一类天然产物中的位置也各不相同,提示自然界中可能存在多种炔基形成机制。
1.1 脂肪酸中炔基的生物合成
炔类脂肪酸是目前分离鉴定数量最多的炔类天然产物,在昆虫[36]、植物[10]与微生物[10]中均有发现。20 世纪60 年代,针对不饱和脂肪酸中炔基的生物合成,研究人员提出了两种可能的机制,分别是氧化脱氢机制与烯醇消除机制。其中,氧化脱氢机制指的是脂肪酸中的顺式双键直接脱氢生成炔基[图1(a)][37]。对于烯醇消除机制,研究人员推测脂肪酸首先经焦磷酸化形成焦磷酸烯醇式中间体, 然后发生脱羧反应生成炔基[图1(b)][38]。

图1 脂肪酸中炔基形成的两种假说Fig.1 Two hypotheses for the biosynthesis of acetylenic bonds in fatty acids
1998 年,Lee 等[39]首先通过体外实验发现高山还阳参(Crepis alpina)种子的微粒体蛋白可以在含有烟酰胺腺嘌呤二核苷酸(NADH)的反应体系中将14C 标记的亚油酸酯(linoleate,1)转化成含有炔基的还阳参油酸酯(crepenynate,2)。而且作者发现当反应体系中加入一氧化碳或P450 还原酶抗体后,依然可以检测到还阳参油酸酯的生成,因此推测催化脂肪酸中炔基合成的酶并非P450 氧化酶,可能是一类与催化脂肪酸中碳碳单键脱氢形成双键的去饱和酶(desaturase)类似的酶。随后,作者从C.alpina种子的cDNA中找到候选基因Crep1,其编码的蛋白与拟南芥中的Δ12 去饱和酶有56%的相似性。最后,作者通过在酿酒酵母以及拟南芥中异源表达证实Crep1 可以将亚油酸酯C-12 与C-13 之间的双键脱氢转化成炔键(图2),并将这类特殊的可以催化炔基生成的去饱和酶命名为乙炔酶(acetylenase)。此后,研究人员又陆续从其他植物、昆虫以及微生物中鉴定了多个乙炔酶(表1)。进一步的研究显示乙炔酶不仅可以将脂肪链中的顺式双键脱氢生成炔基,还可像普通的脂肪酸去饱和酶一样催化双键的形成,如乙炔酶Crep1 可以催化油酸酯(oleate)脱氢生成(Z,E)-9,12-十八碳二烯酸酯[9(Z),12(E)-octadecadienoate]与(Z,Z)-9,12-十八碳二烯酸酯[9(Z),12(Z)-octadecadienoate][40]。
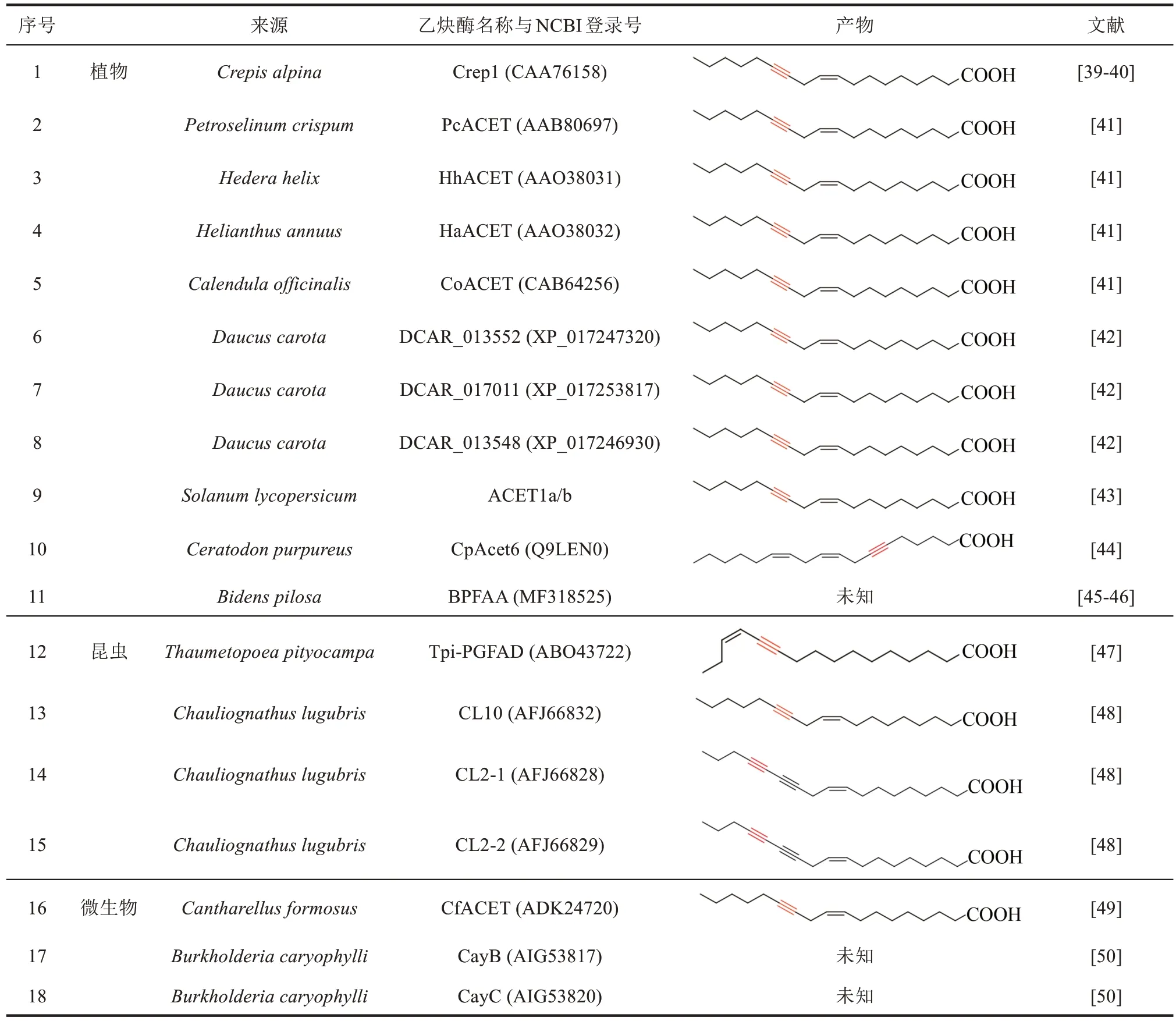
表1 已鉴定的参与炔类脂肪酸生物合成的乙炔酶Tab.1 Acetylenases identified for the biosynthesis of acetylenic fatty acids
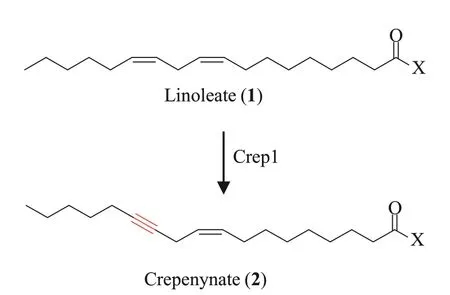
图2 Crep1催化还阳参油酸酯中炔基的形成Fig.2 Crep1-mediated formation of acetylenic bond in crepenynate
虽然研究人员对乙炔酶的认识较晚,但是早在20 世纪60 年代人们已经开始了脂肪酸去饱和酶的研究[51]。去饱和酶是一类在有氧条件下通过二铁活性中心作用于脂肪链碳碳单键的脱氢酶,分为可溶性型和膜结合型[52-54]。其中,可溶性的去饱和酶主要以连接在酰基载体蛋白(ACP)上的脂肪酸为底物,而膜结合的去饱和酶主要作用于与辅酶A、甘油或磷酸连接的脂肪酸。乙炔酶作为一类特殊的去饱和酶也无法直接作用于游离的脂肪酸,而是以脂肪酸酯为底物进行氧化脱氢[39,48,50]。但与普通的去饱和酶不同,目前已鉴定的乙炔酶,包括以连接在酰基载体蛋白CayD 上的脂肪酸为底物的乙炔酶CayB 与CayC[50],均属于膜结合蛋白。2015 年,Bai 等[55]解析了小鼠来源的膜结合型的硬脂酰辅酶A(stearoyl-CoA)去饱和酶SCD1的蛋白晶体结构,发现SCD1 中共有9 个保守的组氨酸负责与活性中心的两个金属铁离子结合,包括H116XXXXH121、 H153XXH156H157、 H294XXH297H298与His265。此外,Asn261 也可通过一分子水结合其中的一个金属离子。将已报道的乙炔酶与SCD1 进行氨基酸序列比较,结果显示乙炔酶中也含有负责与铁离子结合的保守组氨酸残基,此外,有些乙炔酶也如SCD1 一样含有保守的天冬酰胺,但该天冬酰胺在一些乙炔酶中会变为苏氨酸(图3)。此外,CayC 与CpAcet6 仅含有8 个保守的组氨酸残基,另外1个被谷氨酰胺取代(图3),推测这两个乙炔酶可能像可溶性的去饱和酶一样通过谷氨酰胺以及组氨酸与铁离子进行结合[56]。为了阐明乙炔酶催化的碳碳双键脱氢是同步进行还是分步进行,Reed等[57]将2H标记的亚油酸喂养到可以表达Crep1的酿酒酵母中,通过动力学同位素效应实验发现Crep1 首先拔去底物C-12 位上的氢(kH/kD=14.6±3.0),然后C-13 位上的氢再快速离去(kH/kD=1.25±0.8)(图4),这与去饱和酶催化碳碳双键形成的过程类似[58]。

图3 乙炔酶与SCD1的氨基酸序列比对Fig.3 Sequence alignment of acetylenases with SCD1
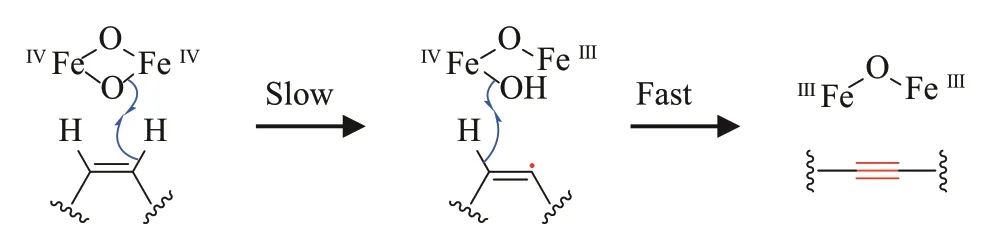
图4 乙炔酶催化机制Fig.4 Proposed catalytic mechanism for acetylenases
1.2 聚酮化合物中内部炔基的生物合成
烯二炔(enediyne)是一类微生物来源的炔类聚酮化合物,其核心骨架中含有两个炔键与同一双键共轭的独特结构。根据核心骨架的差异,此类化合物分为九元烯二炔与十元烯二炔[59-60]。九元烯二炔是由以双环[7.3.0]十二碳烯二炔(bicyclo[7.3.0]dodecadiynene)为核心的发色团与辅基蛋白组成的,其中辅基蛋白主要用于稳定发色团,同时将发色团转运至胞外,如C-1027(3)(图5)。十元烯二炔是以双环[7.3.1]十三碳烯二炔(bicyclo[7.3.1]tridecadiynene)为核心骨架,此类化合物比较稳定,不需要与辅基蛋白结合,如calicheamicin(4)(图5)。不论是九元烯二炔还是十元烯二炔,其核心骨架均可发生重排反应形成苯环双自由基,然后夺取DNA脱氧核糖结构上的氢,从而诱导DNA 双链发生断裂,因此,烯二炔类化合物具有极强的抗肿瘤活性[59-60]。目前,基于多聚物载体系统或抗体偶联的烯二炔类药物已在肿瘤治疗中取得了一定的成功,如聚(苯乙烯-马来酸)-neocarzinostatin于1994年在日本批准上市用于治疗肝癌[61],CD33单克隆抗体-calicheamicin于2000年在美国被FDA批准上市用于治疗白血病[62]。
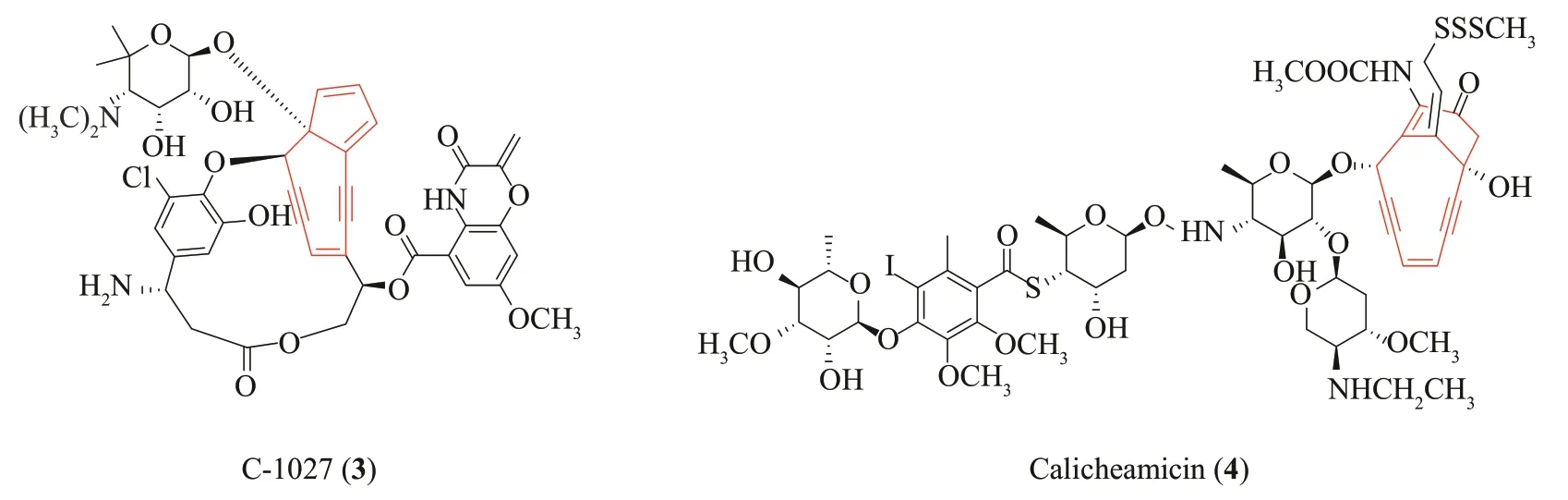
图5 典型的烯二炔抗生素的化学结构Fig.5 Chemical structures of typical enediyne antibiotics
早期关于烯二炔类抗生素的生物合成研究主要采用同位素示踪的方法。Hensens 等[63]通过向neocarzinostatin 生产菌株Streptomyces carzinostaticus喂养13C与14C标记的醋酸钠来探究其生源,结果显示neocarzinostatin的烯二炔核心结构来源于首尾相连的乙酸单元。作者推测其核心结构可能是由油酸酯在脂肪酸乙炔酶的作用下脱氢生成炔基,然后再发生碳碳键断裂以及环化而形成的。Lam等[64]通过向Actinomadura verrucosospora喂养同位素标记的醋酸钠,发现烯二炔esperamicin A1核心骨架上的所有碳原子均来源于醋酸钠,但是作者发现该菌株无法将油酸酯转化成esperamicin A1。尽管同位素标记实验显示烯二炔化合物的核心骨架来源于首尾相连的乙酸单元,但是无法准确判断其到底是来源于脂肪酸途径还是聚酮途径[65]。直到2002 年,Liu 等[66]与 Ahlert 等[67]在同一时间分别找到了C-1027 与calicheamicin 的生物合成基因簇,并通过基因敲除实验发现一类重复使用的Ⅰ型聚酮合酶SgcE 与CalE8 分别参与它们核心骨架的形成。随后,Zhang等[68]发现SgcE以及硫酯酶SgcE10在大肠杆菌中共同表达可催化生成1,3,5,7,9,11,13-pentadecaheptaene(5)(图 6)。同样地,Kong 等[69]通过体外酶催化反应发现CalE8 与硫酯酶CalE7 可催化生成3,5,7,9,11,13-pentadecen-2-one(6)(图6)。由此可见,烯二炔核心骨架的形成还需要其他修饰酶的参与。尽管 Kong 等[69]在 calicheamicin 的生物合成基因簇中发现了一个潜在的去饱和酶基因calU15,但将其与calE8以及calE7一起共表达后并未检测到新的产物。目前,烯二炔抗生素核心骨架中炔基的生物合成机制仍不清楚。
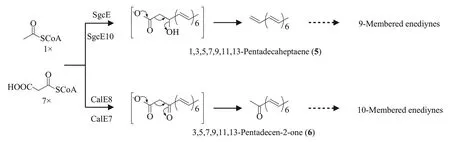
图6 推测的九元与十元烯二炔抗生素的生物合成途径Fig.6 Proposed routes for biosynthesizing 9-membered and 10-membered enediyne antibiotics
1.3 聚酮-非核糖体肽杂合体中末端炔基的生物合成
海洋生物,特别是蓝藻可以合成一类结构特殊的连有末端炔基的肽类化合物[35,70]。2004 年 ,Edwards 等[71]从一株蓝藻Lyngbya majusculaJHB中找到了聚酮-非核糖体肽杂合体jamaicamide B(7)(图7)的生物合成基因簇,发现该基因簇中含有脂肪酰辅酶A 连接酶基因jamA,脂肪酸去饱和酶基因jamB以及酰基载体蛋白编码基因jamC,因此推测JamABC 可能负责jamaicamide B 中末端炔基的合成 。2011 年,Jones 等[72]发 现 carmabin A(8)(图7)的生物合成基因簇中含有与jamABC相似的3 个基因camABC。直到 2015 年,Zhu 等[73]首次阐明了jamaicamide B 中末端炔基的生物合成机制。首先连接酶JamA将5-己烯酸(5-hexenoic acid,9)与酰基载体蛋白JamC 连接形成5-hexenoyl-JamC(10),然后JamB催化5-hexenoyl-JamC中末端的碳碳双键脱氢生成5-hexynoyl-JamC(11)(图8),最后作为起始单元在聚酮合酶以及非核糖体肽合成酶的作用下引入到jamaicamide B 中。此外,作者还对乙炔酶JamB 的底物宽泛性进行了系统研究,发现JamB 对脂肪链的长度,碳碳双键的位置以及酰基载体蛋白JamC 均有严格的要求。为了获得底物特异性不同于JamABC的酶工具,以期将不同长度的含有末端炔基的脂肪链引入到聚酮化合物或聚酮-非核糖体肽杂合体中,Zhu 等[74]以 JamABC 为探针在ⅠMG/JGⅠ数据库中进行了基因挖掘,然后筛选到11 个候选的三基因表达盒。作者通过一系列的体外与体内实验发现来源于Teredinibacter turneraeT7901 的ttuABC可活化 9-癸烯酸 (9-decenoic acid)并将其转化成9-decynoyl-TtuC。

图7 典型的含有末端炔基的聚酮-非核糖体肽杂合体的化学结构Fig.7 Chemical structures of typical terminal alkyne tagged PKS-NRPS hybrids
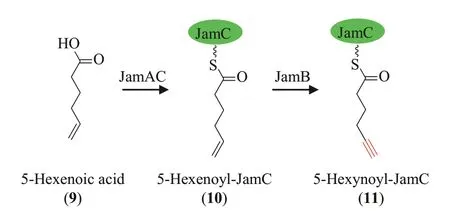
图8 Jamaicamide B中末端炔基的生物合成途径Fig.8 Biosynthetic pathway of terminal alkyne in jamaicamide B
1.4 氨基酸中末端炔基的生物合成
炔类氨基酸在细菌与真菌中均有发现,其炔基既可位于氨基酸侧链的末端,也可位于侧链的内部[75-78]。2019 年,考虑到此前报道的脂肪酸以及聚酮-非核糖体肽杂合体中的炔基均是由一类特殊的去饱和酶(乙炔酶)催化形成的,Marchand等[79]推测 L-β-ethynylserine(βes,12)中的末端炔基可能也是由这类酶催化形成的。但是,作者发现当敲除βes 生产菌株Streptomyces cattleya中的去饱和酶基因后,该菌株仍可合成βes,提示βes中的炔基是由其他酶催化形成的。随后,作者将包括S.cattleya在内的两株可以合成含有末端炔基氨基酸的链霉菌与其他26 株链霉菌进行比较基因组分析,获得了潜在的βes 生物合成基因簇。通过基因敲除与体外酶催化实验,作者阐明了βes 的生物合成途径(图9)。首先,卤化酶BesD 作用于L-lysine(13),在其Cγ位上连接一个氯原子形成4-Cl-L-lysine(14)。然后,氧化酶BesC 催化4-Cl-L-lysine 发生碳碳单键断裂生成4-Cl-allylglycine(15)。裂解酶BesB 通过催化消除反应将4-Clallylglycine 转化成含有末端炔基的 L-propargylglycine(Pra,16)。氨基酸连接酶 BesA 可以将谷氨酸连接到Pra 的氨基上形成二肽化合物γ-L-glutamyl-L-Pra(17),其可在羟化酶BesE 的作用下转化成γ-L-glutamyl-L-βes(18)。最后,在水解酶的作用下,γ-L-glutamyl-L-βes 脱去谷氨酸生成终产物βes。

图9 L-β-ethynylserine的生物合成途径Fig.9 The biosynthetic pathway of L-β-ethynylserine
为了深入了解BesB 是如何催化消除反应形成炔基,作者将4-Cl-allylglycine、BesB 以及辅因子磷酸吡哆醛(PLP)添加至含有氘代水的缓冲溶液中进行反应,最后发现生成的Pra 的分子量增加了2,表明在BesB催化末端炔基形成的过程中发生了两次质子化反应。人们在前期研究Pra 对PLP 依赖的胱硫醚-γ-裂解酶(cystathionineγ-lyase,CSE)的抑制作用时,推测Pra 先与CSE 中的辅因子PLP结合,然后末端的炔基异构成联烯结构,最后再与CSE 中的酪氨酸发生反应从而抑制CSE 的功能[80-81]。因此,作者推测 4-Cl-allylglycine 进入到BesB 的活性口袋后先与PLP 结合形成4-Clallylglycine-aldimine,然后依次脱去Cα与Cβ位上的质子形成4-Cl-allylglycine-ketamine,继续脱去氯离子形成联烯化合物(图10)。该中间体脱去Cδ位上的质子后,再依次在Cβ与Cα位上发生质子化便可形成Pra(图10)。
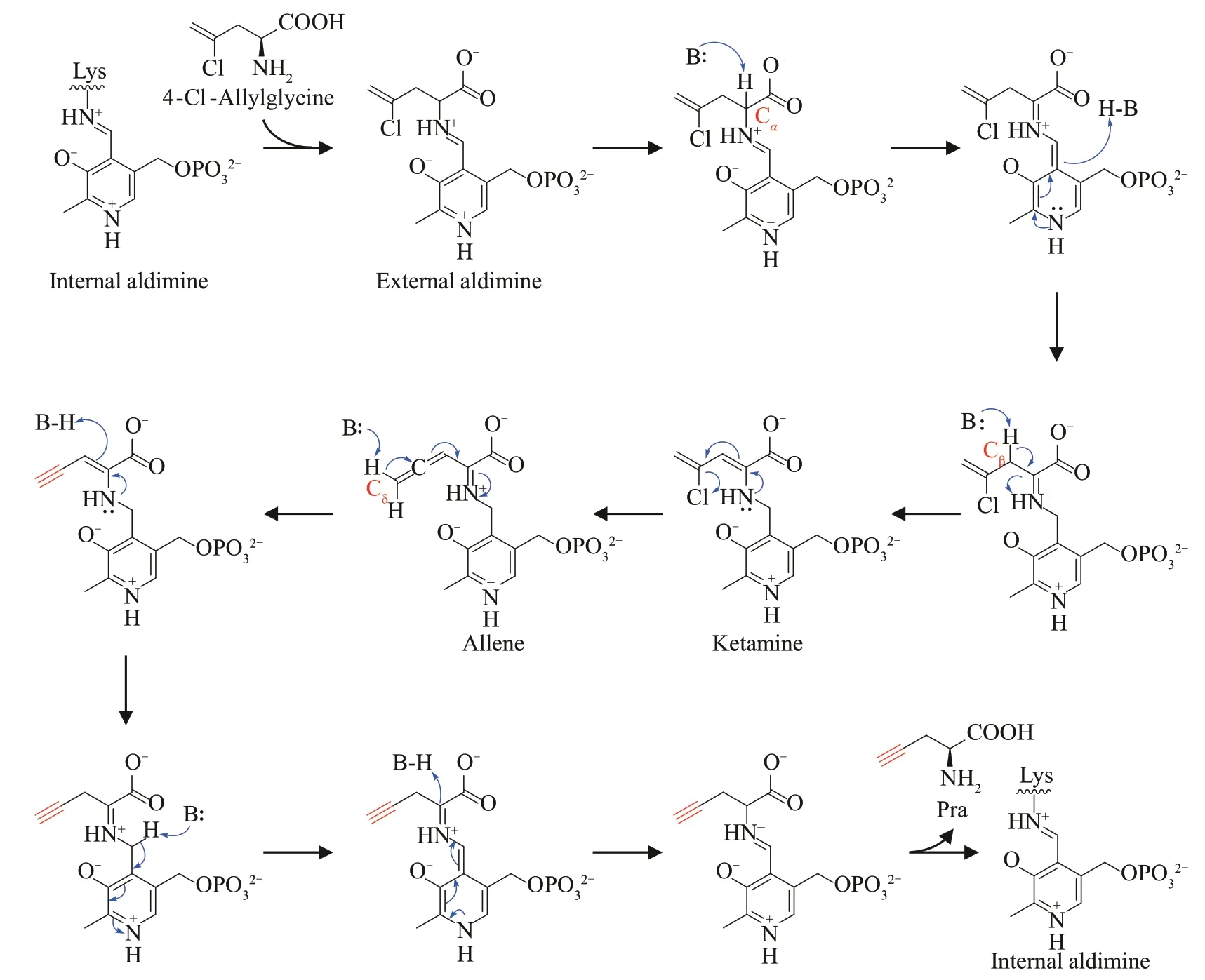
图10 BesB催化的末端炔基形成机制Fig.10 Mechanism for the biosynthesis of terminal alkyne catalyzed by BesB
1.5 杂萜中内部炔基的生物合成
真菌中含有一类特有的异戊烯链中带有炔基的杂萜化合物[8],此类化合物还易与其他化合物发生聚合形成结构更为复杂的聚合物[82-85]。前期,研究人员通过同位素标记实验发现此类化合物的非萜部分来源于莽草酸途径[86],但是其剩余的生物合成途径,特别是异戊烯链中炔基的生物合成机制一直未被阐明。直到2020年,Lü等[87]与Chen等[88]分别解析了炔类杂萜biscognienyne B(19)以及asperpentyn(20)的生物合成途径,发现此类化合物中的炔基是由P450酶催化形成(图11)。
Lü等[87]从一株地衣内生真菌Biscogniauxiasp.(71-10-1-1)中找到了biscognienyne B的生物合成基因簇,并利用米曲霉异源表达体系阐明了biscognienyne B 的生物合成途径(图11)。首先,对羟基苯甲酸(4-HBA,21)在异戊烯基转移酶BisJ的作用下连接一个C5异戊烯链转化成4-hydroxy-3-prenylbenzoic acid(22),其可被P450氧化酶BisⅠ催化生成含有炔基的化合物eutypinic acid(23)。然后,氧化脱羧酶BisD 催化氧化脱羧反应将eutypinic acid 转化成 siccayne(24)。异戊烯基转移酶BisB可连接另一条C5异戊烯链到siccayne上从而生成pestalodiol E(25),最后在含有cupin结构域的氧化酶BisC 与短链脱氢还原酶BisF 的作用下生成终产物biscognienyne B。为了能够检测到BisⅠ催化炔基形成过程中可能存在的中间体,阐明异戊烯链中炔基的形成机制,作者通过化学方法制备了甲基化的底物26,并将其喂养至单独表达BisⅠ的米曲霉转染菌株中。最后,作者从培养体系中分离获得了脱氢产物27 与羟化产物28[图12(a)],提示BisⅠ可能先催化异戊烯链发生脱氢反应生成具有(E)-1,3-diene结构的中间体,然后继续催化反式双键脱氢形成炔基。但是,通过体外反应以及喂养实验,作者发现BisⅠ无法将反式双键转化成炔基。同一时间,Chen 等[88]从Aspergillussp. PSU-RSPG185 中找到了asperpentyn 的生物合成基因簇,包含有UbiA 型异戊烯基转移酶基因atyB,P450 氧化酶基因atyI,氧化脱羧酶基因atyG,核黄素依赖的氧化还原酶基因atyA,含有cupin 结构域的氧化酶基因atyE,短链脱氢还原酶基因atyC,乙醛还原酶基因atyD等,并推测了asperpentyn 的生物合成途径(图11)。通过在酿酒酵母中异源表达,作者证实P450 氧化酶AtyⅠ确实负责asperpentyn 中炔基的形成(图11)。随后,作者合成了一系列的底物类似物,并喂养到单独表达AtyⅠ的酿酒酵母菌株中。喂养结果显示AtyⅠ同样无法作用于反式双键,但是可以将化合物29 中的顺式双键脱氢转化成炔基,从而生成30[图12(b)]。综上所述,P450 氧化酶BisⅠ与 AtyⅠ能够以 4-hydroxy-3-prenylbenzoic acid为底物催化羟化反应与脱氢反应,生成羟基化产物31,以及具有(E)-1,3-diene或(Z)-1,3-diene结构的脱氢产物32 与33,然后再将(Z)-1,3-diene 结构中的顺式双键转化成炔基(图13)。由此可见,P450酶催化的异戊烯链内部炔键的形成机制与去饱和酶催化的脂肪酸链内部炔键的形成机制类似,均是通过脱去顺式双键上的两个氢而形成的,我们推测这是由于底物在酶口袋中无法自由转动,因此酶的金属中心只能拔去双键同侧的两个氢。此外,Lü等[87]发现BisⅠ对底物中异戊烯链的长度没有严格的要求,可识别连有C5或C15异戊烯链的底物。
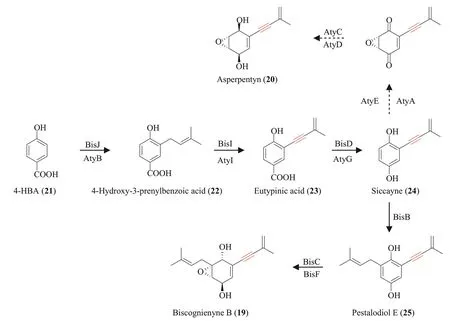
图11 炔类杂萜biscognienyne B与asperpentyn的生物合成途径Fig.11 Routes for the biosynthesis of acetylenic meroterpenoids biscognienyne B and asperpentyn
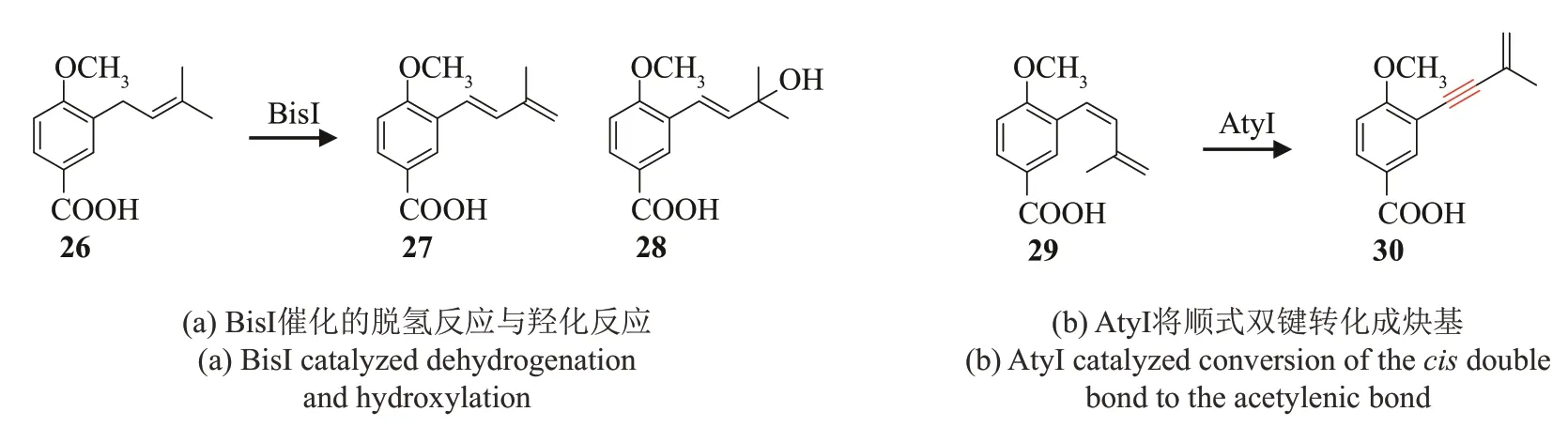
图12 BisⅠ与AtyⅠ功能研究Fig.12 Functional analysis of BisⅠand AtyⅠ

图13 BisⅠ或AtyⅠ催化的炔类杂萜中炔基的生物合成机制Fig.13 Mechanism of alkyne biosynthesis in acetylenic meroterpenoids catalyzed by BisⅠor AtyⅠ
2 非天然炔类化合物的从头生物合成
炔类天然产物的生物合成研究为从头生物合成非天然的炔类化合物提供了必要的酶工具,而且部分炔基合成酶已被用于非天然炔类化合物的构建。
2.1 非天然炔类聚酮的从头生物合成
Zhu等[73]发现jamABC三个基因可以将5-己烯酸转化成5-hexynoyl-JamC(图8),然后作为起始单元被聚酮合酶与非核糖体肽合成酶利用生成jamaicamide B。为了考察jamABC是否可以用于构建含有末端炔基的非天然聚酮化合物,作者将jamABC与Huperzia serrate来源的可以识别不同起始单元的Ⅲ型聚酮合酶基因hspks1[89]一同导入到大肠杆菌Escherichia coliBAP1。由于没有向大肠杆菌中导入5-己烯酸的合成元件,作者在发酵培养基中添加了5-己烯酸,以期jamABC与hspks1可以利用培养基中的5-己烯酸以及大肠杆菌提供的其他原料合成炔基标记的聚酮化合物。经过两天的培养,作者从培养液中检测到了聚酮化合物34(图14),首次实现了非天然炔类化合物的从头生物合成。
Zhu 等[73]和 Porterfield 等[90]还研究了jamABC与Ⅰ型聚酮合酶基因的组合。抗霉素是一类以九元双内酯结构为核心的聚酮-非核糖体肽杂合体,具有多种重要的生物活性[91-92]。前期,研究人员主要通过向培养体系中添加含有炔基的底物来获得炔键标记的抗霉素[29-30]。考虑到抗霉素生物合成途径中的巴豆酰辅酶A 羧化酶/还原酶AntE 以及聚酮合酶AntD 具有宽泛的底物特异性[29],可以引入不同的延伸单元,Zhu 等[73]将antD、antE以及剩余4 个负责抗霉素核心结构生物合成的基因(非核糖体肽合成酶基因antC,酰基辅酶A 连接酶基因antF,酰基载体蛋白基因antG,还原酶基因antM)与jamABC一并导入到E. coliBAP1 中。同样地,由于宿主菌株不含有5-已烯酸与邻氨基苯甲酸的合成元件,作者就在培养基中添加了5-已烯酸与邻氨基苯甲酸,结果显示AntCDEFGM 与JamABC确实可以利用上述原料以及大肠杆菌提供的其他原料合成抗霉素35(图14)。

图14 非天然炔类聚酮的从头生物合成Fig.14 De novo biosynthesis of non-natural acetylenic polyketides
LipPKS1 是脂霉素生物合成的第1 个模块,由6 个结构域组成,包括负责装载起始单元的酰基转移酶(AT)结构域与ACP 结构域,负责装载延伸单元的AT 结构域,酮基还原酶(KR)结构域与ACP结构域,以及负责将起始单元与延伸单元缩合的酮基合酶(KS)结构域[93]。为了将LipPKS1与jamABC进行组合,研究人员移除了LipPKS1 中负责识别与连接起始单元的AT与ACP结构域[90],并将负责以甲基丙二酸单酰辅酶A 作为延伸单元的AT结构域换成可以识别丙二酸单酰辅酶A 的AT结构域,最后在模块的末端增加一个硫酯酶(TE)结构域[94]。Porterfield 等[90]通过体外酶催化反应发现 JamABC 与改造后的 LipPKS1(LipPKS1*)组合后可生成 3-hydroxy-7-octynoic acid(36)(图 14)。
上述研究初步证明负责炔基合成的三基因表达盒jamABC可以与具有宽泛底物特异性的聚酮合酶基因组合从而合成炔类聚酮化合物。基于此,为了实现炔类聚酮化合物的高效从头生物合成,将jamABC与其他所需的聚酮化合物合成元件一起导入到特定的底盘菌株后,一方面可以对三基因合成元件jamABC进行优化[90,95],另一方面也可对底盘细胞与目标化合物合成途径的适配性进行优化[96]。
2.2 非天然炔类蛋白质的从头生物合成
蛋白质中引入连有活泼基团的非蛋白氨基酸在理解蛋白质结构与功能的关系、控制与观察蛋白质在细胞中的定位等方面有重要的应用[97]。早期,人们通过对蛋白质中的半胱氨酸、赖氨酸等进行化学修饰来实现非蛋白氨基酸的引入[98],但是该策略通常只适用于体外分离纯化获得的蛋白,无法用于体内蛋白的修饰[99]。因此,研究人员开发了基因密码子拓展技术,该技术主要是通过对氨酰-tRNA 合 成酶 (aminoacyl-tRNA synthetase)的改造来获得可以识别非蛋白氨基酸的氨酰-tRNA,然后在蛋白质合成过程中,新的氨酰-tRNA 可以将非蛋白氨基酸运输到特定的密码子位点[100]。利用该技术,研究人员已在大肠杆菌、酵母菌以及哺乳动物细胞中实现了连有非蛋白氨基酸的蛋白质的合成[101]。但是,由于此前一直未阐明含有炔基的氨基酸的生物合成途径,研究人员采取直接向培养体系中添加炔类氨基酸来制备炔基标记的蛋白质[97,102]。Marchand 等[79]在阐明了炔类氨基酸βes 的生物合成途径的基础上,将负责中间体Pra 合成的besBCD三个基因,以及经改造的氨酰-tRNA合成酶基因导入到大肠杆菌,即将炔类氨基酸合成模块与新的翻译工具合成模块同时导入到大肠杆菌,以期在不添加炔类氨基酸的情况下利用重组大肠杆菌合成炔基标记的蛋白质。培养两天后,作者将菌株的蛋白粗提物与TAMRA-azide 荧光染料进行点击化学反应,然后通过SDS-PAGE 对反应产物进行分析并用荧光观察,结果发现Pra 确实参与了蛋白质的合成。未来可以对炔类氨基酸合成模块besBCD进行改造优化,拓宽其底物宽泛性,以期获得更多不同的炔类氨基酸,然后利用基因密码子拓展技术合成含有不同炔类氨基酸的蛋白质,为人类在医药、健康等领域的发展提供新的原料。
3 总结与展望
炔基不仅是许多临床药物与活性天然产物的关键功能基因,同时也是在化学反应中被广泛使用的重要结构单元。但是,目前所采用的基于化学合成或底物喂养的炔类化合物合成策略均存在一定的局限性,限制了炔基在药物化学、有机合成以及化学生物学等领域的应用。在合成生物学技术的推动下,从头生物合成被认为是一种可以弥补传统策略的不足,具有广阔应用前景的新的炔类化合物制备技术,其核心要素是需明确天然产物中炔基的形成机制。因此,研究人员开展了一系列炔类天然产物的生物合成研究,以期获得多样的炔基合成酶,为从头生物合成炔类化合物提供酶工具。经过20 多年的努力,人们已从自然界中发现了4种针对不同类型天然产物的炔基形成方式,包括:①在脂肪酸中,乙炔酶催化碳碳双键脱氢形成炔基;②在聚酮-非核糖体肽杂合体中,连接酶先将一条带有末端双键的脂肪酸链与酰基载体蛋白结合,随后乙炔酶将其末端双键脱氢转化成炔基,最后作为起始单元引入到聚酮-非核糖体肽合成体中;③在氨基酸中,卤化酶先催化卤化反应使氨基酸的侧链连接一个卤原子,然后氧化酶使氨基酸的侧链发生断裂生成末端双键,最后裂解酶脱去氨基酸侧链的卤原子从而形成末端炔基;④在杂萜中,P450 氧化酶连续两次催化异戊烯链发生脱氢反应生成炔基。在此基础上,研究人员利用部分炔基合成酶实现了非天然炔类聚酮化合物以及炔基标记蛋白质的从头生物合成。然而,为了更好地满足不同炔类化合物的从头生物合成的需求,研究人员仍需加强多方面的研究工作。
首先,自然界中存在多种结构类型的炔类天然产物,目前仅找到了4种不同的负责炔基合成的基因元件,仍有一些炔类天然产物的炔基形成机制未被阐明,如烯二炔类抗生素核心骨架中的两个炔基以及histrionicotoxin 类生物碱中的炔基等,因此有必要继续开展其他炔类天然产物的生物合成研究,为炔类化合物的生物制造提供种类丰富的酶工具。其次,天然来源的炔基合成酶的底物特异性通常比较严格,无法用于制备结构多样的炔类化合物,因此有必要积极开展炔基合成酶的蛋白晶体结构研究,为拓展其底物特异性提供理论基础。除此之外,炔基合成酶在异源宿主或体外的催化效率问题同样值得关注。尽管上述研究工作仍存在不小的挑战,但是随着基因测序技术、生物信息学技术、基因编辑技术、蛋白晶体学技术等的不断发展,这些瓶颈问题终将会被解决,从头生物合成策略也有望在未来可以广泛地用于炔类化合物的制造,从而与传统的化学合成以及底物喂养策略相互补充,推动炔基在各领域的应用。
猜你喜欢
中学化学(2022年6期)2022-07-05
油气·石油与天然气科学(2021年12期)2021-12-11
商品与质量(2021年31期)2021-11-23
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
疯狂英语·新阅版(2019年6期)2019-09-10
科普创作(2018年1期)2018-11-30
科技资讯(2018年16期)2018-10-26
分析化学(2017年12期)2017-12-25
中学生数理化·高二版(2017年3期)2017-07-07
