
基于相似核融合的多组学数据结直肠癌分子亚型研究*
2021-11-22 07:37武永清郭志飞房瑞玲曹红艳崔跃华
中国卫生统计 2021年5期
武永清 郭志飞 房瑞玲 李 治 曹红艳,4△ 崔跃华
【提 要】 目的 探讨相似核融合(similarity kernel fusion,SKF)方法在整合多组学数据的结直肠癌分子分型中的应用,识别结直肠癌预后高危患者,筛选出潜在的生物标志物以及重要通路。方法 采用SKF对结直肠癌患者多组学数据进行整合,得到融合核,在融合核基础上采用谱聚类进行聚类分型,进一步采用Cox回归研究不同分型患者的预后风险;针对不同分型,筛选差异表达的mRNA(DEmRNAs)、miRNA(DEmiRNAs)以及异常甲基化基因,对三者进行联合分析获得重合基因;最后利用GO和KEGG分析得到重合基因富集的通路。结果 结直肠癌患者分为预后高危组和低危组,其中高危组的死亡风险是低危组的2.72倍,并筛选出1908个DEmRNAs,30个DEmiRNAs,7763个异常甲基化基因,联合分析得到35个基因同时受到mRNA、miRNA及DNA甲基化调控,并富集到有统计学差异的17个GO生物项和23条KEGG通路。结论 SKF能够有效地识别出结直肠癌预后高风险患者,并筛选出结直肠癌的潜在生物标志物及重要通路,为结直肠癌的临床诊断和治疗预后提供重要的思路和依据。
结直肠癌是常见的消化道恶性肿瘤,可以发生在结肠或直肠的任何部位,但以直肠、乙状结肠最为多见。2018年全球肿瘤统计研究显示,结直肠癌的发病率排名第四,占癌症总发病人口的5.7%;致死率排名第二,占癌症总发病人口的9.2%[1]。在我国,结直肠癌的发病率与死亡率都在急剧增长,结直肠癌发病率从 20 世纪 80 年代初的 7/10 万,上升至 2015 年的全国 28.20/10 万;2015年我国因结直肠癌死亡的患者有19.10万人;结直肠癌已成为重大的影响经济的主要公共卫生问题[2-3]。结直肠癌的分子异质性是传统结直肠癌病理不能准确分型的重要原因,对临床分期或病理特征相近的结直肠癌患者采用相同的治疗方案,其治疗反应和临床转归却大相径庭。根据结直肠癌不同生物学行为特征建立其分子分型,以此确定结直肠癌个体化治疗策略,是结直肠癌临床的重要发展方向[4-5]。
结直肠癌分子机制极其复杂,其发生与发展涉及基因变异、表观遗传改变、基因表达异常以及信号通路紊乱等诸多层次的复杂调控机制。随着高通量测序技术的发展,基因组学、转录组学、蛋白组学与代谢组学等多种组学数据海量涌现,如癌症基因组图谱(the cancer genome atlas,TCGA)[6]收集了 33 种癌症相关的20000个癌症样本以及其毗邻的健康组织样本的多组学测序数据,生物医学研究进入了组学时代。任一单组学数据,只能从单一视角反映结直肠癌的内在分子特征,而整合多个组学的信息可以同时捕捉到结直肠癌在不同组学上的异质性[7],以及不同组学之间的关联和潜在的因果关系[8],形成信息互补,从多层面揭示结直肠癌复杂调控机制,有效地提高分型的准确性。
与此同时,整合多组学数据也面临统计建模和计算上的挑战:(1)多个组学的数据加剧了小n大p的“维数灾难”;(2)多组学数据异构,不同组学存在尺度、收集偏倚以及噪声的差异;(3)不同组学数据间互补性信息的处理。Nimrod Rappoport[9]将现有多组学数据整合分析归为五类:①早期集成;②后期集成如PINS;③基于降维的方法如偏最小二乘法;④统计模型法如iCluster;⑤基于相似性的方法如相似网络融合(similarity network fusion,SNF)[10]。其中,基于相似性的方法,分别对每个数据类型构建样本相似矩阵,对不同相似网络进行融合,能够灵活应用于不同类型数据的整合。相似性方法中的SNF采用信息传递(message passing)的方法将多个相似矩阵迭代融合为一个相似网络,无需特征选择,计算速度快,广泛应用于各类癌症的分子分型[11-12]。但是,另一方面,大量噪音信号将稀释SNF分型信号,弱化学习能力。Limin Jiang提出了相似核融合(similarity kernel fusion,SKF)[13]方法,在SNF信息传递迭代算法的基础上,增加了初始相似矩阵项,保留了部分初始相似矩阵信息,同时,对迭代融合核进行降噪,其分型结果更为准确。本文采用SKF算法,整合结直肠癌mRNA、miRNA和DNA甲基化数据,在融合核矩阵基础上采用谱聚类[14]进行分型,识别结直肠癌预后高危人群,并将SKF融合核与单核以及其他整合方法(如SNF和无监督多核学习(unsupervised multiple kernel learning,UMKL)[15]进行比较。针对不同分型,寻找差异临床特征,并寻找不同分型间差异表达基因以及疾病重要通路,从而为结直肠癌的临床诊断、治疗和预后提供新的思路和方法。
数据和方法
1.数据来源
采用TCGA-Assembler[16]从TCGA数据库下载结直肠癌数据,结直肠癌患者总共631人,使用其mRNA表达数据、miRNA数据、DNA甲基化数据及临床数据,进行ID匹配后,得到样本量为336的样本。其中,包含20530个mRNAs,1870个miRNAs以及526729个DNA甲基化位点,结局为生存时间(从确诊到特定事件(死亡或随访结束)发生的时间)及生存状态(随访结束时存活或死亡)。数据预处理方法如下:首先,根据Illumina人源甲基化450芯片平台的注释文件,将甲基化位点转化为其所映射的基因;其次,针对mRNA,miRNA和DNA甲基化数据,删除零表达值所占比例>30%的变量[17],剩余16807个mRNAs,486个miRNAs以及20979个DNA甲基化基因;并对mRNA和miRNAs数据进行log2转换,DNA甲基化数据表达值比较小,因此未进行log2转换。
2.统计分析方法
(1)SKF
SKF在SNF基础上,改进了信息传递迭代算法,并对迭代融合核进行了降噪,其具体步骤和思路如下:
步骤1:对每个数据矩阵构建患者相似核矩阵
(1)
其中xi为第i个患者对应的数据矩阵元素;Ki,j为第i个患者和第j个患者之间的相似性。
(2)
Ni指第i个患者所有邻居的集合。
因此得到mRNA、miRNA以及DNA甲基化数据集的局部相似矩阵Sl(l=1,2,3),Sl∈Rn×n,n为样本例数。
步骤3:对不同组学的相似矩阵进行迭代融合,形成融合核
l=1,2,3
(3)
步骤4:基于融合核Kcom,构建加权矩阵w(i,j)进一步消除融合核的噪声,得到最终的融合矩阵K*,实现对融合核的降噪。
K*=w·Kcom
(4)
(2)谱聚类

minQ∈Rn×c·Trace(QTL+Q)s.t.QTQ=1
(5)
其中,Q=Y(YTY)-1/2为归一化的样本聚类矩阵,D是K*的度矩阵,其对角线元素为对应位置节点的度,非对角线元素设置为0,拉普拉斯矩阵L=D-K*,归一化拉普拉斯矩阵L+=I-D-1/2K*D-1/2,其对角线元素之和为1。谱聚类有效地捕捉了网络图的全局结构。
(3)Cox回归模型
控制年龄、性别、TNM病理分期的情况下,进一步研究结直肠癌不同分型和预后的关系,以生存时间和生存状态(存活或死亡)作为因变量,做Cox回归分析,对结直肠癌的分型进行预后评估。
(4)寻找差异基因
采用DESeq2[19]软件包筛选差异表达的mRNA(DEmRNAs)和miRNA(DEmiRNAs),阈值设定均为Padj<0.001且|log2(foldchange)|>1,利用miRTarBase[20]数据库(http://mirtarbase.mbc.nctu.edu.tw/php/index.php)预测DEmiRNAs的靶基因;采用Limma[21]软件包筛选异常甲基化基因,设定Padj<0.001且|t|>2作为截取标准;通过对DEmRNAs、DEmiRNAs靶基因以及异常甲基化基因进行联合分析,找到重合基因,并利用KOBAS[22]在线工具对重合基因实现GO[23]和KEGG[24]富集分析。SKF采用Matlab R2018b软件进行分析,其余分析采用R3.5.3软件。
结 果
1.SKF整合与单个组学及其他整合方法分型结果比较
多组学数据整合的相似矩阵图相比于单一数据源,具有明显的块状结构,同时,SKF的聚类图像相比于SNF和UMKL更为清晰,如图1。进一步比较不同方法的分型在生存预后的差异,做log-rank检验,基于单个组学数据的分型在生存状况上无显著差异,SKF、SNF和UMKL不同分型在生存率上均存在显著差异,但SKF分型优于SNF及UMKL,如图2。结果表明SKF能够捕获不同组学数据的共同信号和互补信号,其分型结果相比单个组学和SNF、UMKL分型更为准确。

图1 单个数据和数据整合后的相似矩阵图
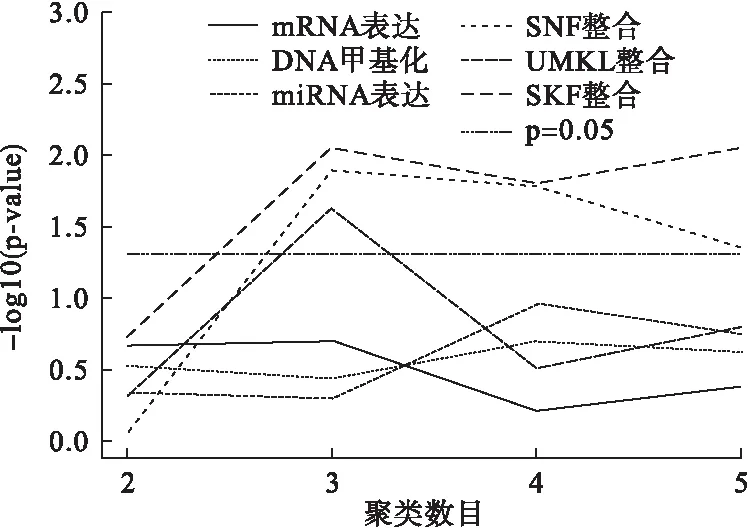
图2 单个数据和数据整合后的聚类数与对应的P值关系图
2.SKF参数选择
α是SKF整合过程的重要参数,较小的α表示在融合核中保留更多的初始信息。设置α从0到1,增长梯度为0.1,寻找结直肠癌数据最适合的α值。结果如图3,在α=0.8,聚类结果为3类时,得到结直肠癌的最优P值,因此后续研究选择α=0.8。

图3 参数α与其对应的P值关系图
3.基于SKF的结直肠癌患者分型结果评价
采用SKF的方法对336名结直肠癌患者进行组学数据整合分型,在聚类结果为3的情况下,P值最小(图2),因此,将结直肠癌患者分为3组。Group 1患者为164例(48.8%),3年死亡率为18.6%;Group 2患者为103例(30.7%),3年死亡率为56.6%;Group 3患者为69例(20.5%),3年死亡率为27.0%。Cox检验发现Group 1和Group 3生存率没有统计学差异(χ2=0.124,P=0.725),因此,将Group 1和Group 3合并同时命名为低危组,Group 2命名为高危组,两组基本资料如表1,生存曲线如图4,生存曲线显示每个时间点低危组的生存率均高于高危组,说明分型有一定的临床意义。
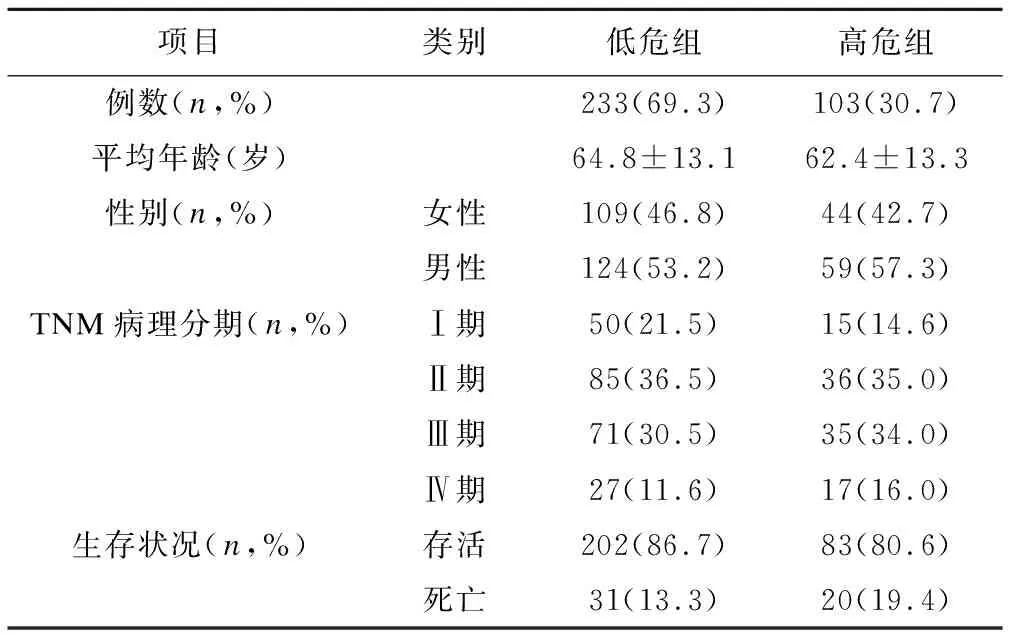
表1 结直肠癌患者分型的基本资料
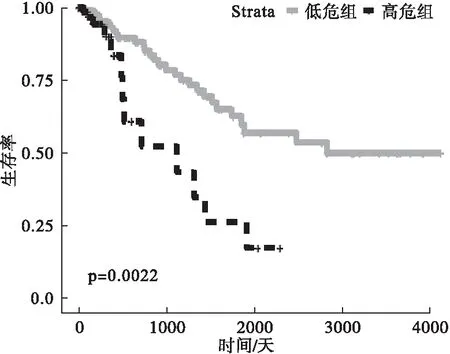
图4 低危组与高危组的生存曲线图
校正协变量年龄、性别及TNM病理分期的情况下,研究不同分型对预后的影响,即分型作为自变量,生存时间和生存状态(存活或死亡)作为因变量,拟合Cox回归模型。回归结果如表2所示,在校正协变量的情况下,高危组患者的死亡风险是低危组的2.722倍;年龄和TNM病理分期均有统计学意义,随着年龄的增加,结直肠癌患者的死亡风险增加;TNM病理分期是结直肠癌患者的死亡危险因素。
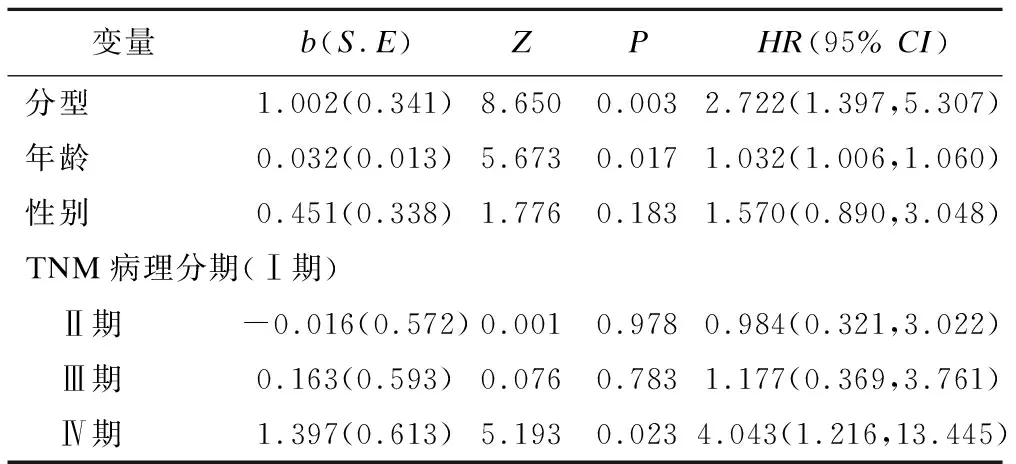
表2 336例结肠直肠癌患者多变量Cox回归分析结果
4.低危组与高危组差异基因分析
(1)差异基因的筛选结果
以低危组为对照组,筛选在两种分型之间差异表达基因。筛选出1908个DEmRNAs,其中上调1872个,下调36个;筛选出7763个异常甲基化修饰基因,包括3648个甲基化修饰水平升高的基因和4115个甲基化修饰水平降低的基因;筛选出30个DEmiRNAs,其中17个上调,13个下调。图5依次为DEmRNAs、异常甲基化基因及DEmiRNAs热图,其中行为特征,列为样本,从图中可以明显看出这些特征在低危组与高危组中有显著差异的表达情况。

图5 DEmRNAs、异常甲基化基因、DEmiRNAs在不同分型的表达热图
(2)DEmRNA、DEmiRNA和异常甲基化基因的联合分析
通过MiRTarBase预测,鉴定出30个DEmiRNAs的1073个靶基因。通过对1908个DEmRNAs,7763个异常甲基化基因以及1073个DEmiRNAs靶基因进行联合分析,发现35个基因既mRNA差异表达,同时异常甲基化且对应的miRNAs差异表达(图6)。其中AKAP12低表达,且CpG岛高甲基化,对应的miRNAs高表达;CLEC14A,CLIP4,FGF2,FGF5,FNBP1,GPR21,HSPB2,MYL9,MYO5A,PPP1R18,RSPO1,SEMA7A,SLFN11,SOGA3,SOX11,SYNM,TAL1,TM4SF1,VEGFC,ZNF154这20个基因高表达,且CpG岛低甲基化,对应的miRNAs低表达,值得进一步研究。
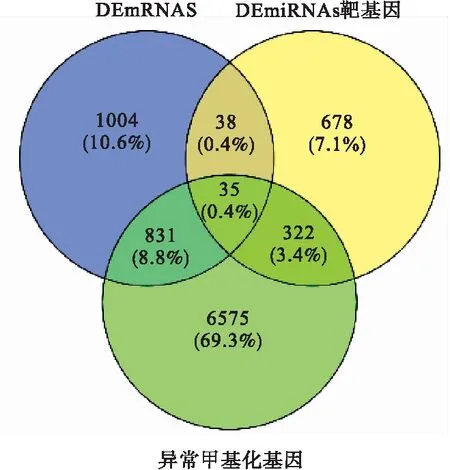
图6 DEmRNAs、DEmiRNAs靶基因及异常甲基化基因的关系
(3)生物功能注释结果
为了进一步验证SKF得出的结直肠癌亚型的生物学意义,本文选择了联合分析结果中35个重合基因进行GO富集分析以及KEGG通路分析。35个基因富集于17个adj-pvalue<0.05的GO生物过程项,如图7;KEGG通路分析中,一共有23条adj-pvalue<0.05的通路被定位,见图8。
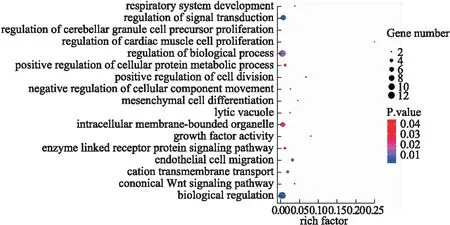
图7 GO通路分析图

图8 KEGG通路分析图
讨 论
本文针对结直肠癌多基因组学数据,采用SKF进行整合分型,将结直肠癌患者分为高危组和低危组,高危组患者的死亡风险是低危组的2.72倍,同时识别出差异基因并找到结直肠癌的重要通路,有效地实现了结直肠癌的分型,识别出高危患者,为结直肠癌患者进一步有的放矢地治疗提供了方法参考和重要的生物标志物。
高危组和低危组的显著差异分子靶标中,35个基因同时受到mRNA、miRNA和DNA 甲基化多方面调控,其中,CLEC14A,CLIP4,FGF2,FGF5,FNBP1,GPR21,HSPB2,MYL9,MYO5A,PPP1R18,RSPO1,SEMA7A,SLFN11,SOGA3,SOX11,SYNM,TAL1,TM4SF1,VEGFC,ZNF154这20个基因高表达,且CpG岛低甲基化,对应的miRNAs低表达,值得进一步研究;另外,多项研究已证实受到DEmiRNAs负调控的高甲基化基因AKAP12可抑制结直肠癌细胞的增殖、侵袭、趋化和新生血管形成[25-26]。TM4SF1,VEGFC高表达与结直肠癌的深部侵袭、淋巴结转移有关[27-28];本研究SLFN11发生低甲基化高表达,应属于致癌基因,但有研究报道SLFN11启动子附近在结直肠癌、胃癌中经常发生甲基化,SLFN11能够抑制结肠癌细胞的增殖[29-30]。除此之外,本研究发现的结直肠癌致癌基因CLEC14A与肿瘤的发展有很强的联系,被认为是有吸引力的癌症治疗候选靶点,RSPO1是影响胃癌患者预后的独立危险因素,以及FGF2,FGF5在胶质母细胞瘤和乳腺癌中的过表达已被报道[31-35]。
本研究发现结直肠癌富集的17个GO生物项和23条KEGG通路。GO生物项代表的生物过程大部分都与癌症的产生发育和转移有着密切联系。其中最为显著的是Wnt经典信号通路,研究表明,该通路的异常活化影响结直肠癌的发生发展和转移[36];除此之外,间质细胞分化也可能促进恶性肿瘤细胞的生长,内皮细胞迁移通过影响肿瘤血管的生成促进肿瘤生长和转移[37-38];其他项包括生长因子活性调节、细胞分裂正调控等都是参与结直肠癌整个生命周期的重要生物过程[39-40]。KEGG通路中,可以看出所有的通路之中癌症相关是最主要的子类,例如癌症通路、肿瘤坏死因子信号通路、癌症中的蛋白多糖、癌症中的转录失调。除了直接的癌症通路,Ras信号通路的激活会促进结直肠癌细胞增殖[41];MAPK信号通路的激活会增强肠道炎症,促进结直肠癌的发展[42];PI3K-AKT信号通路促进结直肠癌细胞增殖,并且与结直肠癌的侵袭和转移密切相关[43]。
本研究中,随着年龄的增加,结直肠癌患者预后逐渐变差[44];TNM病理分期为结直肠癌患者死亡的影响因素,TNM分期越高,患者预后越差[45]。
SKF分型在融合核基础上采用谱聚类进行分型,当聚类维度非常高时,谱聚类运行速度较慢且聚类效果不够理想,今后将改进现有的谱聚类算法,以提升算法速度及聚类的准确性。另外,本研究找到的潜在生物标志物,还需进一步的分子生物学验证。
总之,本文基于结直肠癌的多基因组学数据的SKF整合分型,充分利用了患者mRNA、miRNA和 DNA甲基化的信息,识别出结直肠癌预后高危患者,为结直肠癌的分型研究提供了新的思路和方法,筛选出的潜在生物标志物及重要通路,对结直肠癌的临床治疗和预后具有重要的指导意义。
猜你喜欢
中国医学影像技术(2021年2期)2021-03-30
国际口腔医学杂志(2019年3期)2019-05-31
天然产物研究与开发(2018年2期)2018-04-04
实验与检验医学(2017年3期)2017-06-27
医学研究杂志(2015年11期)2015-06-10
癌变·畸变·突变(2015年3期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
遗传(2014年3期)2014-02-28
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
中国全科医学(2014年22期)2014-02-08
