
生物质气再燃脱除流化床N2O 的机理研究
2021-11-20 08:35:04牛胜利王永征韩奎华周文波
燃料化学学报 2021年10期
李 颖 ,牛胜利 ,王永征 ,韩奎华 ,周文波 ,王 俊
(山东大学 能源与动力工程学院, 山东 济南 250061)
近年来,循环流化床(CFB)锅炉因具有较高的燃烧效率,较低的脱硫设备投资、运行成本及污染物排放量而受到广泛应用[1]。循环流化床的NOx生成量明显低于常规的煤粉炉,但是其释放的N2O 体积量可达到一般锅炉的100 倍以上[2,3]。作为《京都议定书》规定的六种温室气体之一,N2O 被认为是21 世纪最主要的臭氧消耗物质[4,5],因此,较高的N2O 排放量已成为制约CFB 锅炉发展的瓶颈。再燃技术是控制流化床燃煤过程中N2O 排放的最常采用的方法[6−8]。生物质能具有生态意义上碳的零排放,同时其硫、氮含量较低,在燃煤锅炉中采用生物质燃料再燃的方式,不仅能够控制生物质能的利用规模,还可以减少常规化石燃料燃烧过程中硫氧化物和氮氧化物的排放[9]。生物质通过气化炉气化产生的以CO、H2和CH4为有效成分的燃料气中碱金属及氯元素含量低[10],可以克服生物质直接燃烧引起的锅炉受热面的灰腐蚀,以及炉灰玷污严重等问题[11,12],是理想的再燃用燃料。
CaO 作为CFB 锅炉中常用的脱硫剂及循环灰的主要成分,在其炉膛和气固分离系统中大量存在。目前,普遍认为CaO 有助于减少CFB 燃煤过程中N2O 的排放。侯祥松等[13]通过探究石灰石脱硫对循环流化床N2O 排放的影响,发现减排作用主要是通过石灰石受热分解产生的CaO 对N2O的分解的催化作用所致。周浩生等[14]实验研究了CaO 及CO 对N2O 的催化还原作用,结果表明,在CaO 和CO 的多相催化和均相反应共同作用下,能够有效地降低N2O 分解的活化能。胡笑颖[15]通过实验研究了CFB 电厂循环灰中CaO对生物质气还原分解N2O 的过程,认为CaO 对N2O 的抑制作用主要表现为对N2O 的直接分解(式(1))和由CO参与的间接分解的催化作用(式(2)),且在CaO 的作用下N2O 分解率随着生物质气含量的增加而增大。因此,利用现有CFB 锅炉中CaO 成分较多的特点,结合生物质气再燃技术,是实现N2O 较高脱除率的有效途径。
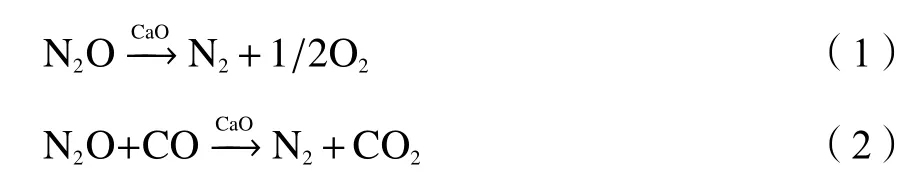
基于密度泛函理论(density functional theory, DFT)的量子化学研究是计算化学领域的常用方法,能在埃纳尺度层面上描述催化剂的过程本质,有助于系统、深入地揭示催化机理。因此,基于前人的实验研究结果,有研究人员从微观反应机理角度出发研究了CaO 催化N2O 的反应机理[16−18]。Hu 等[19]结合实验,利用量子化学的方法研究了N2O 分子在CaO(100)表面的吸附,认为CaO(100)表面对于N2O 分子的分解有很高的催化活性。Snis 等[20]通过实验及量子化学研究了CaO 及MgO 对N2O 分解的催化活性,发现CaO 具有更高的催化活性。吴令男[21]结合实验,基于密度泛函理论研究了N2O在CaO 表面的非均相分解路径,计算结果表明,N2O 在CaO(100)表面向N2的转化可以自发进行,但向NO 的转化不能自发进行。综上,已有实验表明CO 的加入能够促进CaO 对N2O 脱除的催化,但目前微观理论的研究大都集中于N2O 在CaO 表面的吸附及分解情况,而有关生物质气再燃过程中CO 对CaO 催化N2O 的脱除影响的反应机理研究不足。
本研究基于密度泛函理论(DFT)和经典过渡态理论(TST),针对CFB 锅炉N2O 排放量偏高的特点,借助分子模拟的手段,设计了不同的反应路径,探究了CFB 锅炉内生物质气再燃过程对CaO催化N2O 脱除的影响。本文所得结论能够为控制CFB 锅炉高效燃烧过程中N2O 的排放控制提供理论指导。
1 计算方法和模型
本研究所有计算均利用Dmol3模块完成,采用自旋非限制广义梯度(Generalized Gradient Approximation)耦合Perdew-Burke-Ernzerhof泛函的方法(GGA-PBE)。计算精度在进行反应物、过渡态、中间体、产物的几何结构优化时设置为Medium(energy: 2 × 10−5Ha;Max force:0.04 Ha/nm; Max displacement: 5 × 10−4nm;1 Ha = 2625.5 kJ/mol),自洽场(SCF)的总能量收敛极限为1.0 × 10−5Ha,在过渡态搜索(transition state,TS)时设置为Coarse(RMS convergence: 0.2 Ha/nm)。采用加极化函数展开的双数值基组(DNP)处理价电子波函数,核心电子使用Effective Core Potential处理,同时计算采用Tkatchenko-Scheffler 方法进行DFT-D 修正,所有计算均考虑自选非限制性(Spin:unrestricted),且将热拖尾效应(smearing)设置为0.005 Ha 加速收敛。使用频率振动分析证明各优化结构的合理性,结果表明,产物、中间体及产物无虚频,过渡态存在唯一虚频。同时,采用相同计算水平对优化后的结构进行单点能计算,并在此基础上进行零点能校正,本文中所有计算均基于校正后的能量进行。
氧化钙是典型的立方晶系结构,具有Fm-3 m(No.225)空间点群。采用上述参数优化得到的CaO 晶胞参数为0.482 nm,与实验测量值基本一致[22]。选取CaO 晶体最易暴露且表面能最低的CaO(100)表面为研究对象[23],采用p(2 × 2)的五层周期模板,计算时固定底层三层原子使其保持CaO 体相结构,其余原子层自由松弛。为消除周期结构之间的相互作用,相邻两层平面的真空层设置为1.2 nm。
表面吸附能Ead定义为吸附前后各物质总能量的变化,吸附能越大表示吸附越稳定。吸附能的表达式如式(3)所示。

式中,Eads和Esub分别表示吸附前吸附质底物的能量,Esys表示吸附后整个体系的总能量。其中,Ead为负值时表示吸附过程为放热反应,该值越小则吸附作用越强烈。
2 结果与讨论
2.1 N2O 的均相分解
N2O 在大约600 ℃以上才会发生如式(4)所示的均相热分解反应[24]。

N2O 热分解过程的反应物(R)、过渡态(TS1)及产物(P)的优化结构如图1 所示,相应的反应势能面如图2 所示。其中,N2O 的N1−O1键逐渐伸长,具体值为0.1194 nm(R)→0.1974 nm(TS)→0.2809 nm(P),最终导致N1−O1键断裂。该过程需要克服346.782 kJ/mol 的能垒,并且需要吸收282.285 kJ/mol的能量,最终生成N2分子和O 自由基。

图1 N2O 的均相分解反应各驻点结构示意图Figure 1 Structures of stagnation points of N2O homogeneous decomposition (Bond length: nm)
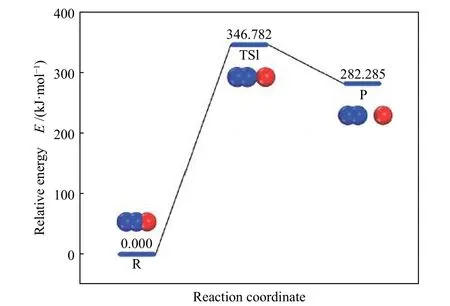
图2 N2O 的均相分解反应势能面Figure 2 Reaction potential energy surface of N2O homogeneous decomposition
2.2 CaO(100)表面N2O 的非均相分解
2.2.1 N2O 在CaO(100)表面的吸附
由前文可知,N2O 在CaO 表面会发生如式(1)所示的非均相反应。本文首先探讨了N2O 分子在CaO(100)表面O 位点及Ca 位点的吸附结构,最终得出了图3 所示的四种稳定吸附构型。其中,N2O 分子O1端在CaO(100)表面O 位点及Ca 位点吸附释放的能量分别为−83.575 及−33.715 kJ/mol,N2端在CaO(100)表面O 位点及Ca 位点吸附释放的能量分别为−15.129 及−42.977 kJ/mol。对比吸附能大小可知,N2O 分子更倾向于以O1端吸附在CaO(100)的O 位点,与Snis 等[25]的研究结果一致。电子态密度可用于解释吸附分子与吸附表面的电子相互作用[26]。如图4所示,对不同吸附情况的电子态密度进行了讨论以研究吸附后N2O 分子的吸附端原子和CaO(100)表面吸附位点的电子特性。由4(b)、(c)、(d)可知,N2O 分子吸附在Ca(100)表面后,其吸附端原子与表面位点均没有发生轨道杂化,表示N2O 分子与Ca(100)表面的弱相互作用。而由图4(a)可知,N2O 分子以O1端吸附在CaO(100)表面的O 位点后,吸附的原子O1与表面O 位点发生了明显的杂化作用,两者的2s轨道在−22、−17 及−14 eV 处出现轨道重叠,2p轨道在−6、−4 eV 及费米能级附近存在轨道重叠,反映了原子O1与O 位点之间的共价键作用。电子态密度计算结果与吸附能分析结果一致。最优结构如图5 所示,N2O 分子的N1−O1键断裂,原子O1吸附于表面O 位点,距离为0.1505 nm;裂解生成的N2分子键长为0.1109 nm,与自由N2分子一致;原子O1与N1的距离为0.2862 nm。因此,N2O分子的O 端在CaO(100)表面O 位点上吸附后导致了N2O 分解为吸附的原子氧及自由的N2分子。

图3 N2O 分子在CaO(100)表面吸附的稳定构型Figure 3 Stable adsorption structures of N2O on the CaO(100) surfaces (Bond length: nm; blue: N atom; red: O atom; green: Ca atom)

图4 四种稳定构型在CaO(100)表面吸附后的局部态密度图Figure 4 Partial densities of states of the four stable structures after adsorption on the CaO(100) surfaces(a) O-O, (b) O-Ca, (c) N-O, (d) N-Ca
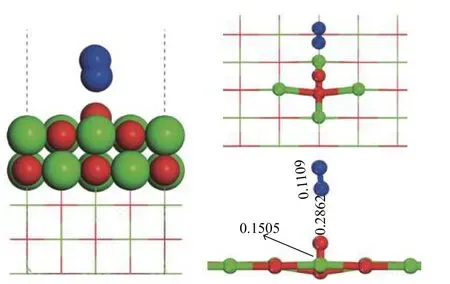
图5 N2O 分子在CaO(100)表面吸附的最优构型Figure 5 Optimal adsorption structure of N2O on the CaO(100)surface (Bond length: nm)
2.2.2 N2O 在CaO(100)表面的分解过程
图6 为N2O 分子在CaO(100)表面分解过程中的反应物(R)、过渡态(TS1)及生成物(P)的优化结构,其反应势能面如图7 所示。N2O 分子物理吸附在CaO(100)表面的O 位点后,N1−O1键逐步伸长断裂,变化数值为0.1197 nm(R)→0.1794 nm(TS1)→0.2862 nm(P),O1与表面O 位点逐渐接近,变化数值为0.3067 nm(R)→0.2519 nm(TS1)→0.1505 nm(P)。最终N2分子脱附,O1原子与表面O 位点成键。该反应过程需要克服190.907 kJ/mol能垒并释放31.855 kJ/mol 能量。对比N2O 的非催化均相热分解,CaO 可以显著降低N2O 中N1−O1键断裂所需克服的能垒,且吸附构型更加稳定,从而促进N2O 的分解。
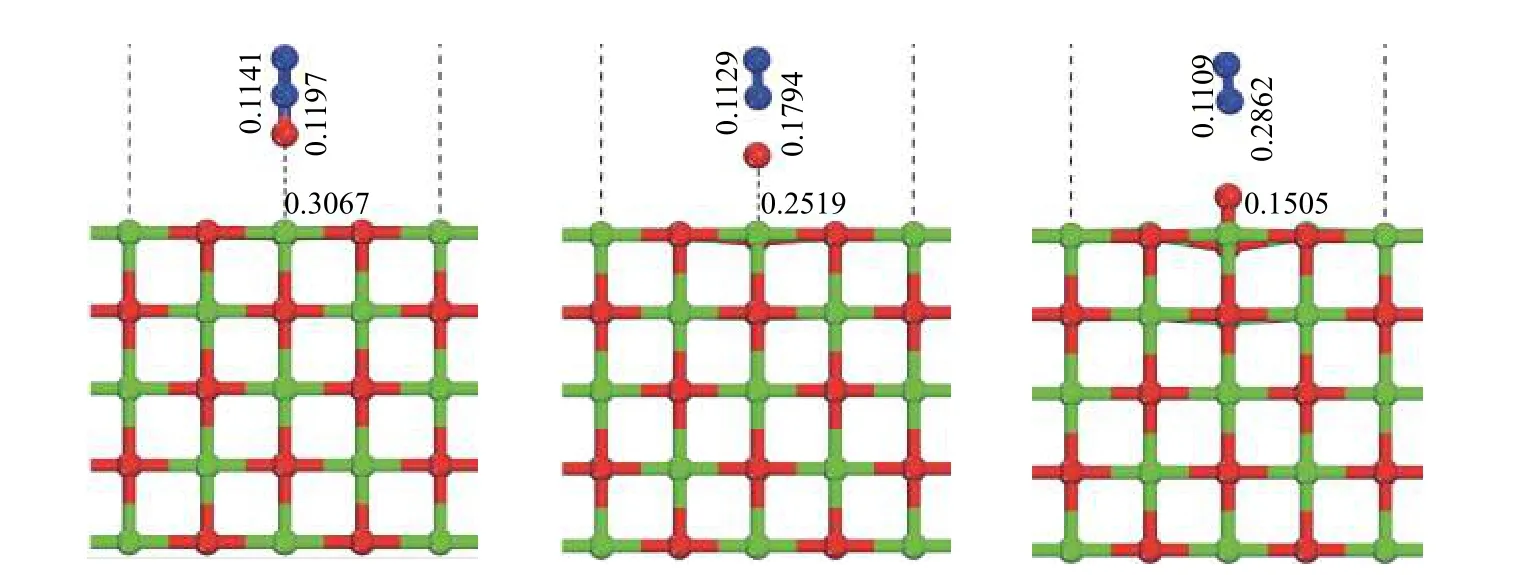
图6 CaO(100)表面N2O 的分解反应各驻点结构示意图Figure 6 Structures of stagnation points of N2O decomposition on the CaO(100) surface (Bond length: nm)

图7 CaO(100)表面N2O 的分解反应势能面Figure 7 Reaction potential energy surface of N2O decomposition on the CaO(100) surface
Mulliken 电荷布局分析反映了吸附体系中原子与键在吸附前后的电子分布变化[27]。对N2O 分子O1端在CaO(100)表面O 位点吸附前后进行了Mulliken 电荷布局分析。发现吸附后CaO(100)表面的O 原子相较于纯CaO(100)表面电负性变小,由−1.246 e 变为−0.829 e;而N2O 分子的O1原子电负性变大,由−0.279 e 变为−0.649 e,表明表面O 位点与O1之间发生了电荷转移。即在CaO(100)表面O位点的催化作用下,N2O 的O1原子通过N1−O1键的活化与断裂过程及其与表面O 位点之间的成键过程,由N2O 分子脱离并吸附至CaO(100)表面的O 位点,形成过氧根基团,同时气态N2分子脱附。
2.2.3 CaO(100)表面的再生
由前文可知,N2O 分子在CaO(100)表面热解后,其O 原子占据了CaO(100)表面的O 位点。若吸附O 原子过多,则表面活性催化O 位点被高度覆盖,因此探究了O 原子的表面吸附对CaO 催化N2O 分解的影响。Fu 等[28]认为O 原子吸附在表面后,后续吸附的N2O 将通过式(5)及式(6)的反应对CaO 表面进行还原,从而实现催化剂的表面再生。

进一步,图8 给出了N2O 在CaO(100)表面吸附的O 原子分解过程中的反应物(R)、过渡态(TS1、TS2)、中间体(M)及生成物(P)的优化结构,相应的反应势能面如图9 所示。首先N2O 分子以O1端吸附于表面吸附态的O 原子处,N2O 分子的O1端与吸附态的O 原子逐渐靠近(0.3294 nm(R)→0.2501 nm(TS1)→0.1359 nm(M))并成键,而其N1-O1键逐步断裂(0.1192 nm(R)→0.1940 nm(TS1)→0.3062 nm(M)),该过程克服了291.047 kJ/mol的能垒并需要吸收17.471 kJ/mol 的能量。随后O2及N2分子从表面脱附,CaO(100)表面还原为初态,需克服75.145 kJ/mol 的能垒同时释放40.838 kJ/mol能量。

图8 N2O 参与的CaO(100)表面再生各驻点结构示意图Figure 8 Structures of stagnation points of the recovery process on the CaO(100) surfaces with N2O (Bond length: nm)

图9 N2O 参与的CaO(100)表面再生反应势能面Figure 9 Reaction potential energy surface of the recovery process on the CaO(100) surfaces with N2O
因此,该反应过程的最大能垒存在于表面O原子的脱附过程,说明该步为反应的决速步骤[29]。虽然N2O 对Ca(100)表面的还原过程所需能垒低于N2O 的均相裂解,但明显高于N2O 在Ca(100)表面的分解过程,这与Wu 等的结论一致[30]。Leglise等[31]通过实验发现N2O 在催化剂表面的脱除过程中,N2O 对催化剂表面的还原是主要的速率限制步。因此,随着N2O 在CaO(100)表面的分解,表面O 位点覆盖的吸附态O 原子增多,CaO(100)表面的活性位点被逐渐钝化,明显降低了CaO 对N2O分解的催化活性。
2.3 生物质气再燃时CaO(100)表面N2O的分解过程
2.3.1 CO 在CaO(100)表面的吸附
张磊等[32]认为,CaO 对于由CO 参与的N2O均相还原反应通过式(2)有显著的催化作用。本文首先考虑了CO 与N2O 分子在CaO(100)表面上的竞争吸附。CO 在CaO(100)表面上的稳定吸附结构如图10 所示,与N2O 分子的吸附相似,包括CO 的C 端及O 端在Ca 位点与O 位点的吸附四种结构。其中,CO 分子的C 端在CaO(100)表面的O 和Ca 位点的吸附能分别为−61.365 和−70.193 kJ/mol,O 端在O 和Ca 位点的吸附能分别为−41.532 和−68.008 kJ/mol。相比于CO 分子,N2O分子在CaO(100)表面上的吸附能更大,因而将会优先吸附在CaO(100)表面并发生分解。
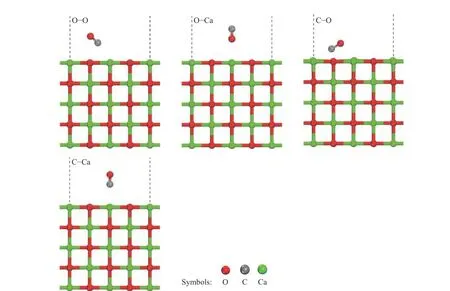
图10 CO 分子在CaO(100)表面吸附的稳定构型Figure 10 Stable adsorption structures of CO on the CaO(100) surfaces (gray: C atom; red: O atom; green: Ca atom)
2.3.2 CaO(100)表面的再生
Kapteijn 等[33]认为,当存在CO 时,N2O 分解后的催化剂表面的原子O 脱除步骤如式(7)所示。

CO 对CaO(100)表面还原过程中的反应物(R)、中间体(M)、过渡态(TS1、TS2)及生成物(P)如图11 所示,图12 则为该过程的反应势能面。根据2.3.1,N2O 分子在CaO(100)表面吸附并发生分解,生成自由N2分子和一个原子O。CO分子随后吸附在CaO(100)表面,并逐渐靠近吸附于表面O 位点的O 原子,CO 分子中的C 原子与O 原子间距缩短(0.2411 nm(R)→0.1695 nm(TS1)→0.1260 nm(M)),最终C 原子与O 原子成键。原子O 则远离吸附位点(0.1508 nm(R)→0.1931 nm(TS1)→0.2282 nm(M)),此过程克服了151.511 kJ/mol的能垒,且放出大量热(409.841 kJ/mol)。最后,M 吸收115.141 kJ/mol 的能量形成了自由的CO2分子且从CaO(100)表面脱附,该过程克服的能垒为127.591 kJ/mol。由反应过程可知,其反应决速步也为表面原子O 的脱附步。由此可知,CO 对原子O 吸附的Ca(100)表面的还原过程所需能垒远低于N2O 对Ca(100)表面的还原过程,且低于N2O在CaO(100)表面的分解能垒。Debbagh 等[34]实验发现,CO 对催化剂表面吸附原子O 的去除效果比通过N2O 更为显著,这与本文计算结果一致。因此,生物质气CO 促进了CaO 表面O 活性位点的快速还原,有利于N2O 在CaO 表面的吸附及CaO表面对N2O 的分解的催化。

图 11 CO 参与的CaO(100)表面再生各驻点结构示意图Figure 11 Structures of stagnation points of the recovery process on the CaO(100) surface with CO (Bond length: nm)

图12 CO 参与的CaO(100)表面再生反应势能面Figure 12 Reaction potential energy surface of the recovery process on the CaO(100) surface with CO
3 结 论
相 比 于N2O 的 均 相 分 解,N2O 在CaO(100)表面分解所需的能垒更低,即CaO 的存在有利于N2O 的脱除。
随着N2O 在CaO(100)表面的分解,CaO(100)表面O 活性位点逐渐被吸附的原子O 钝化,不利于后续N2O 的分解。
生物质气再燃过程CO 的存在促进了CaO(100)表面吸附的原子O 的去除和CaO(100)表面的还原,有利于CaO 对N2O 分解的催化作用。
利用循环流化床锅炉N2O 排放量大及CaO 含量多的特点,结合生物质气再燃技术,是减少循环流化床锅炉N2O 排放的重要途径。
致 谢
本论文的科学计算得到了山东大学的高性能计算云平台的计算支持和帮助。
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
能源工程(2021年5期)2021-11-20 05:50:44
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
生物质化学工程(2021年1期)2021-01-26 09:22:30
中国造纸(2020年9期)2020-10-20 05:33:36
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24

- 燃料化学学报的其它文章
- Catalytic pyrolysis of sugarcane bagasse by zeolite catalyst for the production of multi-walled carbon nanotubes
- Nitrogen-doped porous carbon supported nickel nanoparticles as catalyst for catalytic hydroconversion of high-temperature coal tar
- Probing into the crystal plane effect on the reduction of α-Fe2O3 in CO by Operando Raman spectroscopy
- CoSOH/Co(OH)2 复合纳米片的制备及其氧析出催化性能
- Effects of promoters on carburized fused iron catalysts in Fischer-Tropsch synthesis
- Effects of Ca content on the activity of HZSM-5 nanoparticles in the conversion of methanol to olefins and coke formation