
环境空气暴露对猪粪悬液中菌群及代谢物的影响
2021-11-18 08:08孙静陈奕龄丁玉春葛良鹏
中国实验动物学报 2021年5期
孙静陈奕龄丁玉春葛良鹏*
(1.重庆市畜牧科学院,重庆 402460;2.农业部养猪科学重点实验室,重庆 402460;3.重庆市养猪科学重点实验室,重庆 402460;4.重庆市医用动物资源开发与利用工程技术研究中心,重庆 402460)
目前,粪菌移植(fecal microbiota transplantation,FMT)已成为干预微生物组的主要手段之一。微生物组的可变性为FMT等治疗性干预措施提供了希望,但也可能引起了严重的安全隐患。由于肠道微生态系统的复杂性高,无目的地干预措施可能会将微生物组转移为不期望的状态,反而给健康带来更意想不到的后果。比如,抗性基因的传播[1-2]、产气荚膜梭菌感染[3]等不良事件的发生。供体的选择、供体粪便的处理方式、受试者的准备、移植方式和次数等都可能影响FMT的疗效。目前,用于复发性CDI治疗的FMT并无统一的标准和监管,不同国家的政策差别较大[4]。在丹麦,Jørgensen等[5]采取了比较严格的供体筛选方法,包括病史、体格检查、血液、粪便以及尿液检查,仅约20%的参与者符合捐赠粪便资格。Ramai等[6]根据PubMed、MEDLINE、Google Scholar和Cochrane数据库上FMT相关研究,推论证实FMT的成功并不取决于供体-患者的关系;Allegretti等[7]对1924例FMT治疗艰难梭菌感染(Clostridioides difficileinfection,CDI)的数据进行分析,发现粪便运输时间和粪便冻存时间对FMT疗效的影响差异不明显:粪便处理时长超过150 min,并未显著降低FMT的疗效;当粪便冻存时长超过1年甚至3年时,FMT的疗效与冻存30 d内的效果相似。
同样是杂食动物,猪与人在消化道解剖学和生理学特征方面具有相似性。最近多项研究还利用无菌猪模型实现了动物肠道菌群“拟人化”的能力。比如,Zhang等[8]对无菌仔猪接种成人和婴儿的粪便悬液,成功模拟了成人和婴儿的肠道微生物群落结构;接种患有严重急性营养不良和中度急性营养不良的儿童的粪悬液构建悉生猪模型,揭示了共生肠道菌群与宿主健康生长存在因果关系,阐明了治疗儿童营养不良的方法[9]。此外,肠道微生物组与宿主动物的共同进化,它在营养消化与利用、抵御病原体、调节免疫和内分泌等方面发挥的微生物作用,促使养猪生产对FMT这一策略的应用也产生了兴趣。比如,通过比较常规饲养猪和悉生猪或无菌猪的生理和代谢参数等来研究肠道微生物的作用[10];或使用FMT作为提高生猪饲料利用率的可能策略等[11]。Hu等[12]以金华猪为例,尝试利用FMT方式调节杂种新生仔猪肠道菌群结构,来改善肠道屏障和免疫功能。这些研究提示FMT在构建悉生猪模型上的应用广泛,那么,对影响FMT的相关因素开展研究就十分必要。供体选择、粪便样本的处理、粪菌移植方式、粪菌库建立以及安全性保障等是影响FMT实施和效果的几个关键因素。本研究以SPF猪作为供体,在有氧或厌氧下处理并制备得到粪便悬液,比较环境空气暴露对粪便悬液中微生物组和代谢产物的影响。
1 材料与方法
1.1 材料
1.1.1 实验动物
供体猪由重庆市畜牧科学院双河实验猪基地自繁获得【SCXK(渝)2018-0002】。在符合现行国标(GB 14925-2010)规定的屏障环境内饲养,自由采食和饮水【SCXK(渝)2018-0002】。参考GB/T22914-2008《SPF猪病原的控制与监测》标准,选择健康的5头成年雌性无特定病原级别(specific pathogen free,SPF)巴马小型猪(耳号分别为13,14,69,77,78),体重约50 kg,包括非洲猪瘟病毒、猪瘟病毒、口蹄疫病毒、猪繁殖与呼吸综合征病毒、乙型脑炎病毒、猪圆环病毒2型、猪细小病毒、布鲁氏菌、猪肺炎支原体、猪流感病毒、猪弓形虫、猪流行性腹泻病毒、猪传染性胃肠炎病毒、轮状病毒在内的13种病原均为阴性。经重庆市畜牧科学院伦理委员会的许可(XKY-20150113),所有实验均在重庆市畜牧科学院重庆国家现代畜牧业示范区实验用猪工程中心完成(重庆荣昌)【SYXK(渝)2018-0002】。
1.1.2 供体猪粪便的收集
供体猪选定后,单栏饲养。采样当天清晨,打扫其圈舍至无明显残留粪污。采用无菌密封采样袋分别收集上述5头供体猪的新鲜粪便,每头各收集3份。其中1份直接放入液氮保存(未处理对照组,或称C组),1份直接保存在低温运输箱(4℃)(有氧暴露组,或称T1组),剩余1份转入厌氧采样盒(由密封培养罐和厌氧培养应用袋制造厌氧环境,广东环凯微生物科技有限公司,中国),再保存于低温运输箱(4℃)(厌氧暴露组,或称T2组),在30 min内运回实验室进一步处理。
1.1.3 主要试剂与仪器
QIAamp DNA stool Mini Kit(QIAGEN公司);AxyPreDNA凝胶回收试剂盒(AXYGEN公司);TruSeqTM DNA Sample Prep Kit;色谱柱为BEH C18柱(100mm×2.1mm i.d.,1.7μm;Waters,Milford,USA)。厌氧工作站(英国DWS公司,型号DG250);洁净工作台(北京东联哈尔仪器制造有限公司,型号DL-CJ-2ND1);QuantiFlourTM-ST蓝色荧光定量系统(美国Promega公司);PCR仪(美国ABI,GeneAmp9700型);高分辨液质联用仪UHPLC-Q Exactive HF-X系统(赛默飞公司)。
1.2 方法
1.2.1 供体猪粪悬液的制备
猪供体粪便的处理上参考Pang等[13]描述的方法。T1组在超净台内(氮气约占78%,氧气约为21%)完成粪悬液的制备,T2组在厌氧工作站内(纯氮气和无氧混合气体供应,氮气>80%,其余气体为氢气和二氧化碳)完成粪悬液的制备。制备过程在样品采集后2 h内完成。粪悬液的制备操作为:无菌密封匀浆袋内,新鲜粪便和灭菌水按照1∶5的比例混合均匀,再通过4层灭菌医用纱布过滤,收集获得粪悬液。每头供体猪的新鲜粪便样品(约2 g),各个粪悬液样品抽取4 mL,用于细菌基因组的提取和非靶向代谢组分析;剩余粪悬液和灭菌甘油按照体积比9∶1混合,分装成每管1 mL,-80℃保存。
1.2.2 测序与分析
(1)扩增子测序:采用CTAB/SDS方法提取各粪便样品的总基因组DNA,然后用338F/806R引物扩增16S rRNA的V3-V4区域,ITS1F/ITS2R引物对扩增真菌ITS区域。利用Illumina Miseq测序平台完成扩增子测序,微生物多样性和组成的确定参考之前的分析方法[14]。
(2)非靶向代谢组:采用LC-MS分析平台(赛默飞公司,超高效液相色谱串联傅里叶变换质谱UHPLC-Q Exactive HF-X系统)对15个猪粪便样品进行代谢组学分析。样品经过前处理去除杂质、提取代谢物,然后LC-MS正、负模式下分别上级检测采集信息,得到代谢物的MS和MS/MS信息,采用Progenesis QI(Waters Corporation,Milford,USA)软件进行搜库鉴定,将MS和MS/MS质谱信息与代谢数据库(http://www.hmdb.ca/;http://metlin.scripps.edu/)进行匹配,最终得到代谢物列表及数据矩阵。
1.2.3 数据分析
细菌和真菌功能预测分析:利用Tax4Fun对16S RNA基因序列进行COG和KEGG功能注释,获得OTU在COG和KEGG各功能水平的注释信息及各功能在不同样本中的丰度信息。FUNGuild用于真菌功能分类分析。
1.3 统计学分析
Student’st检验用于比较α多样性(Shannon指数和Chao指数)的组间差异;单因素方差分析用于比较组间微生物丰度差异;Heatmap分析展示不同分组/样品在各分类学水平上的群落组成的相似性和差异性;组间差异检验采用ANOSIM分析方法;基于Bray-Curtis距离的PCoA分析展示各组样品之间的微生物总体分布和离散程度;PCA用于观察各组样品之间的代谢物的总体分布和离散程度;PLSDA用于区分组间差异,变量重要性(variable importance,VIP)>1用于筛选出对模型贡献较大的变量。本研究以VIP>1的变量作为差异代谢物或潜在标志物。采用Student’st检验结合OPLS-DA方法,筛选出组间差异代谢物(同时满足VIP>1,P<0.05)。Spearman相关系数r展示菌群组成与差异代谢物丰度上的相关关系。
2 结果
2.1 细菌组成与丰度比较
利用338F/806R引物对,对C、T1和T2这三组共15个样品中微生物群落测序,共获得raw reads 645275*2,总碱基数目为388 455 550;优化后有效序列数目为645 275,有效碱基数目为266 910 646;序列平均长度为413.64 bp(见表1)。
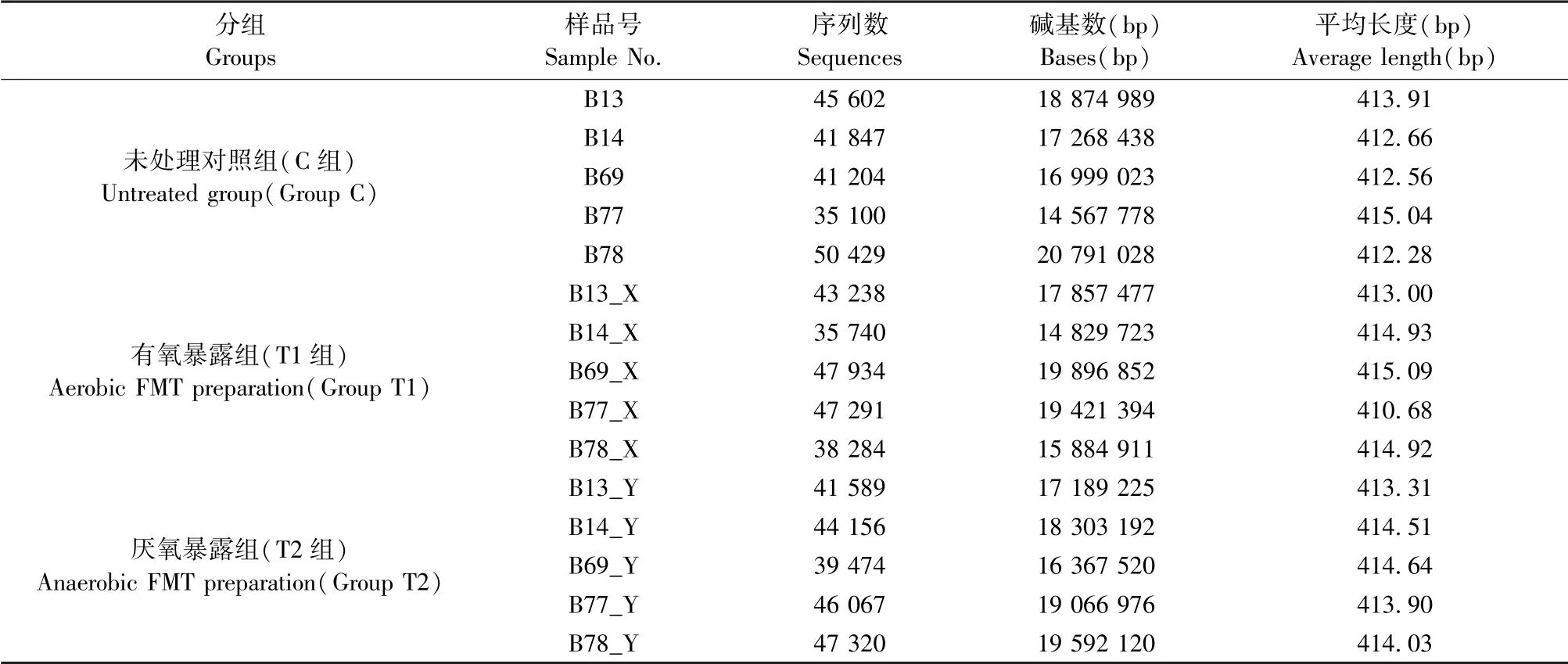
表1 基于Illumina Miseq测序平台的16S rRNA V3-V4测序结果Table 1 Illumina Miseq sequencing of the V3-V4 region of the bacterial 16S rRNA gene
对97%相似水平下的细菌OTUs进行统计分析,15个样品中共检出1020个OTUs。C组、T1组和T2组检出的OTUs数目分别为915、982以及968,其中856个OTUs在三组间共享。此外,FMT制备操作(T1或T2组)与C组在Shannon指数和Chao指数上组间差异显著(P<0.05,图1A,1B),提示需氧或厌氧下供体猪粪便的FMT制备操作均提高了样品中菌群多样性。相似性分析(ANOSIM检验)显示R值为0.314(P=0.005),表明组间差异大于组内差异,提示实验分组成立。PCoA分析显示T1与T2组在猪粪便菌群结构上组间差异小,而与C组间差异明显(图1C)。
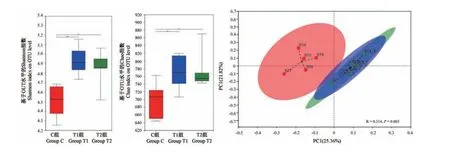
图1 FMT制备处理对SPF级猪粪便样品中细菌群落多样性的影响Note.A.Student’s t test for Shannon index of OTU level.B.Student’s t test for Chao index of OTU level.C.PCoA analysis.*P<0.05,**P<0.01.Figure 1 Influence of FMT preparation on bacterial community diversity in SPF pigs’fecal samples
FMT制备操作保存了新鲜供体粪样中的全部细菌门,组间共享18种细菌门(图2A)。其中,厚壁菌门(Firmicutes)的相对平均丰度在C、T1和T2组中分别为65.07%、62.39%和61.90%,其次为拟杆菌门(Bacteroidetes),它在C、T1和T2组中相对平均丰度分别为23.98%、32.72%和33.06%。FMT制备操作后,大部分细菌门的丰度组间差异不明显(P>0.05),仅显著改变了黏胶球形菌门(Lentisphaerae)和互养菌门(Synergistetes)丰度(P=0.035和P=0.002)。两者在C组中的相对丰度极低,在T1组的相对平均丰度分别为0.109%和0.037%,T2组中分别为0.019%和0.040%。值得注意的是,尽管螺旋体门(Spriochaetes)的丰度在组间差异不显著(P>0.05),但需氧下FMT制备操作后丰度从8.34%降低到1.28%,厌氧下降低为1.13%。该菌在C组各样本中丰度变化较大,从2.18%~13.28%。这提示,FMT制备操作对猪粪便中的Spirochaetes丰度影响较大。Heatmap分析显示T1和T2组菌落先聚类,再和C组聚类(图2B)。
T1和T2组间细菌属组成上无明显差异,但丰度上差异明显。相对丰度较高的细菌属主要集中在Firmicutes和Bacteroidetes,比如Lchnospiraceae_XPB1014 group,Prevotellaceae_NK3B31 group,Ruminococcaceae UCG_005。和C组相比,T1和T2组的Terrisporobacter,Treponema_2的丰度显著低于C组(P<0.05),而Ruminococcaceae_UGC_005,Rikenellaceae_RC9_gutgroup以及Ruminococcaceae_NK4A214 group的丰度显著高于C组(P<0.05)(图2C,2D)。
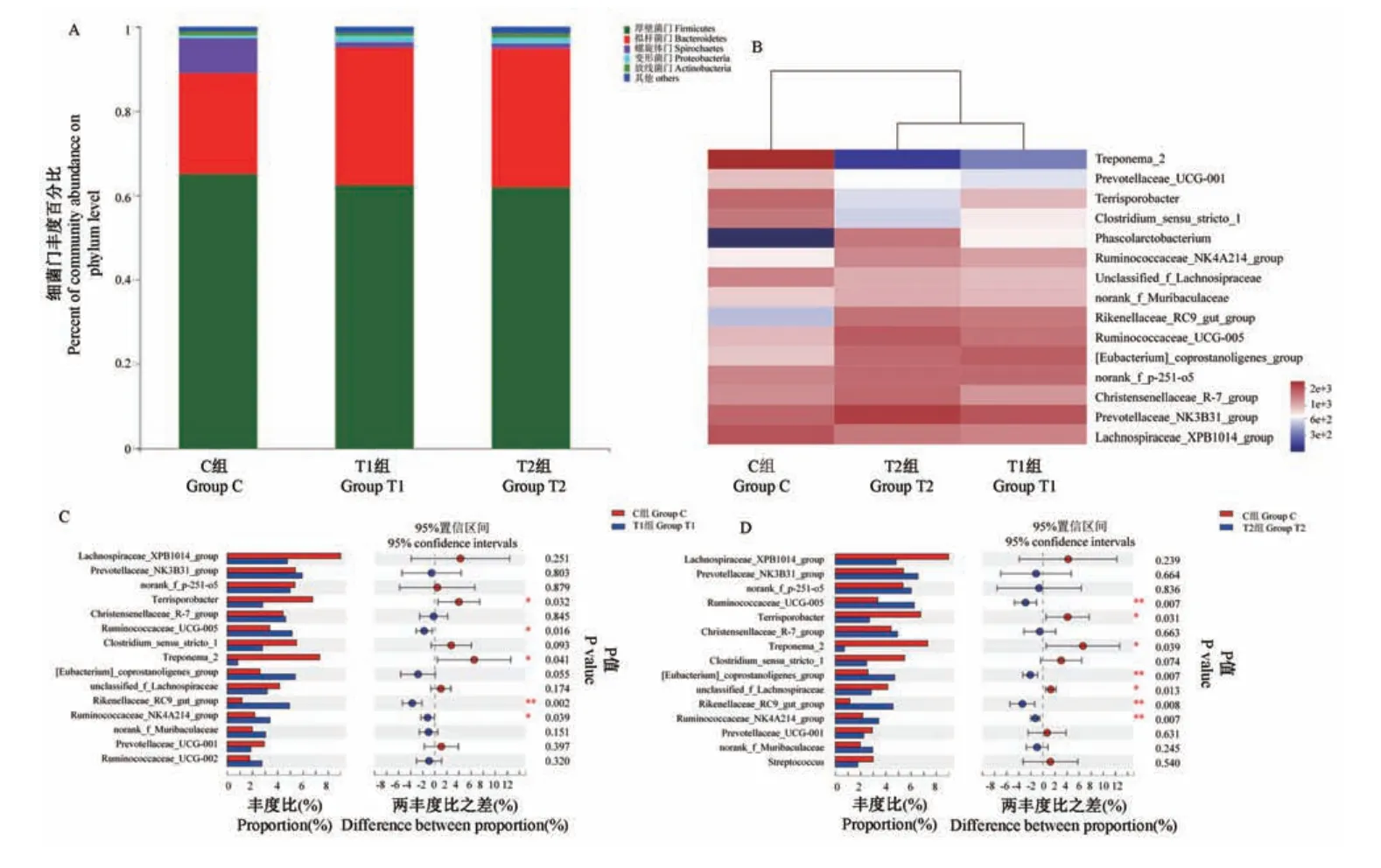
图2 FMT制备处理对供体猪粪便细菌群落组成的影响Note.A.Barplot results of community abundance on bacterial phylum level.B.Heatmap results on bacterial genus level.C.Student’s t-test barplot on bacterial genus level between C and T1 groups.D.Student’s t-test barplot on bacterial genus level between group C and T2.*P<0.05,**P<0.01.Figure 2 Influence of FMT preparation on bacterial community of SPF pigs’feces
2.2 真菌组成和丰度比较
利用ITS1F-ITS2R引物对,对各样品中真菌群落组成情况进行测序。结果显示raw reads数为898449*2,总碱基数目为540 866 298,有效碱基数目为245 604 522,具体测序信息见表2。

表2 基于Illumina Miseq sequencing平台的真菌ITS1-ITS2区域的测序结果Table 2 Illumina Miseq sequencing of ribosomal RNA ITS1-ITS2 genomic region in fecal mycobiota
对97%相似水平下的真菌OTUs进行统计分析,15个样品中共检出158个OTUs。C组、T1组和T2组各检出的OTUs数目分别为128、85和77个,其中3组间共享48个OTUs。Shannon指数组间差异不显著(P>0.05);C组与T1组、T2组之间在Chao指数上差异显著(P<0.01)。ANOSIM检验显示R=-0.068,P>0.05,提示猪粪便中真菌组成,组间差异不明显,与PLS-DA分析结果相同(见图3)。

图3 FMT制备操作对供体猪粪便样品中真菌群落多样性的影响Note.A.Student’s t-test for Shannon index of OTU level.B.Student’s t-test for Chao index of OTU level.C.PLS-DA analysis.**P<0.01.Figure 3 Influence of FMT preparation on the fungal community diversity in SPF pigs’fecal samples
C组共识别出128个真菌OTU,T1组识别出85个,T2组识别出77个OTU。群落组成分析结果显示,C组以子囊菌门(Ascomycota)为主,相对平均丰度为63.26%;其次为担子菌门(Basidionmycota)和Neocallimastigomycota,分别占27.13%和9.17%。当粪便经过FMT制备操作处理后,无论是在需氧或厌氧条件下Ascomycota相对丰度大幅度提升,在T1组平均丰度达到87.65%,在T2组达到85.15%;而Neocallimastigomycota丰度降低到1%以下(T1组为0.25%,T2组为0.44%)。基于样本中群落丰度数据,结果显示在真菌门上组间差异均不显著(P>0.05);节担菌科(Wallemiaceae)、Sporidiobolaceae和Bulleribasidiaceae组间存在显著差异(P<0.05),这三种真菌科在C组的丰度很低,少于1%。Wallemiaceae在C组的平均丰度为0.999% ±1.049%,在T2组中丰度为0.476%±1.065%,但在T1组未检出。在真菌属上,节单菌属(Wallemia)的丰度在组间存在差异,但它在C组和T2组中丰度也低于1%,在T1组中未检出(图4)。这提示这类真菌在需氧下FMT制备操作时,可能因为其含量过低而未被保存下来,而厌氧暴露似乎部分保存了这类真菌。

图4 FMT制备处理对供体猪粪便真菌群落组成的影响Note.A.Barplot results of community abundance on fungal phylum level.B.One-way ANOVA barplot results of community abundance on fungal family level.C.One-way ANOVA barplot results of community abundance on fungal genus level.*P<0.05.Figure 4 Influence of FMT preparation on fungal community of SPF pigs’fecal samples
2.3 菌群功能预测
利用Tax4Fun对样品中细菌进行COG和KEGG pathway功能注释和丰度比较,结果显示共注释到24个COG功能分类,组间丰度相似。其中,Carbohydrate transport and metabolism和Amino acid transport and metabolism丰度最高,相对丰度均超过8.0%(P>0.05,图5A)。KEGG pathway level1水平上共注释到 cellular processes,environmental information processing,genetic information processing,human diseases,metabolism以及Organismal systems这6个KEGG通路,组间差异小(P>0.05)。其中,注释到metabolism的通路达到137个,但各相对丰度在组间差异不明显(P>0.05)。
FUNGuild通过微生态guild对真菌群落进行分类分析。由图5B发现,粪便中真菌的主要功能为腐生营养型,其中未定义腐生真菌(undefined Saprotroph)平均相对丰度在C组高达77.56%,T1组为87.92%,T2组为83.53%。C组中,Animal Endosymbiont-Plant Saprotroph平均相对丰度达到9.89%,略高于Animal Pathogen的平均相对丰度(7.35%)。相比之下,粪便经过过滤处理后,Animal Endosymbiont-Plant Saprotroph丰度急剧减少,在T1组仅为0.34%,T2组为0.85%;其他功能分类,如Animal Pathogen,组间丰度差异不明显(P>0.05)。

图5 T1、T2和C组中粪菌功能预测结果Note.A.The result of COG function classification of fecal bacterial community from donor pigs.B.The result of fungal functional annotation from donor pigs inferred by FUNGuild.Figure 5 Bacterial and fungal function classification in groups C,T1 and T2
2.4 代谢组分析
2.4.1 数据质控
利用LC-MS分析平台测定15个猪粪便样品中代谢物组成和丰度情况,并采用PCA对数据结果进行质控分析。质控样本(Quality Control,QC)用于评价在上级过程中分析系统的稳定性。QC样本相关性越高(越接近于1)表明测试方法稳定性越好,数据质量越高。在正负离子2个模式下,QC样本相关性最小值为0.994,表明此次检测方法稳定,数据可信(见图6)。通过PLS-DA得到模型评价参数(R2,Q2),R2和Q2值越接近1,表明模型越稳定可靠;如果R2和Q2<0.5,则模型可靠性较差。相关评价参数结果:C组与T2组比较时(即T2:C代谢集):R2X=0.513,R2Y=0.935,Q2=0.897;C组与T1组比较时(即T1:C代谢集):R2X=0.521,R2Y=0.614,Q2=0.451;说明模型构建稳定,数据可靠;T1与T2比较时(即T2:T1代谢集):R2X=0.326,R2Y=0.41,Q2=-0.709,提示T1和T2模型效果不好,这两组间得到的差异物质不可信。
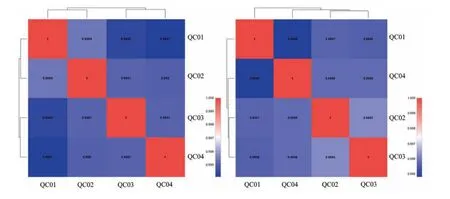
图6 QC样本相关性分析结果Note.A.Positive ion mode.B.Negative ion mode.Figure 6 Results of QC sample correlation analysis
2.4.2 代谢物分类统计
利用代谢组数据库(HMDB,www.hmdb.ca)对猪粪便中代谢物进行注释。正、负离子模式下,402和195种代谢物被分类为14个超类别(superclass)和21个子类别(subclass)。其中,有243种代谢物注释到“脂类和类脂分子”(lipids and lipid-like molecules),有54种代谢物注释到“有机杂环化合物”(organoheterocyclic compounds),50种代谢物注释到“有机酸和杂环化合物”(organic acid and derivatives)。子类别中,注释到“氨基酸、肽和类似物”(amino acids,peptides and analogues),“脂肪酸和偶联物”(fatty acids and conjugates)以及“胆汁酸、醇类和衍生物”(bile acids,alcohols and derivatives)的代谢物数量分别为41、26和25个。
2.4.3 差异代谢产物的筛选与鉴定
采用Student’st检验结合OPLS-DA,筛选出组间差异代谢物。同时满足变量重要性VIP>1,P<0.05,差异倍数FC>1或FC<1。阳离子模式下T1-C代谢集筛选出差异代谢物数量为90个,T2-C代谢集筛选出差异代谢物数量122个,T2-T1代谢集筛选出差异代谢物3个。阴离子模式下T1-C代谢集筛选出差异代谢物65个,T2-C代谢集筛选出差异代谢物79个,T2-T1代谢集筛选出差异代谢物1个(见表3)。因此,T1:C代谢集筛选出的差异代谢物共有155个,T2:C代谢集筛选出的差异代谢物共有201个,它们之间共享的差异代谢物达142种。
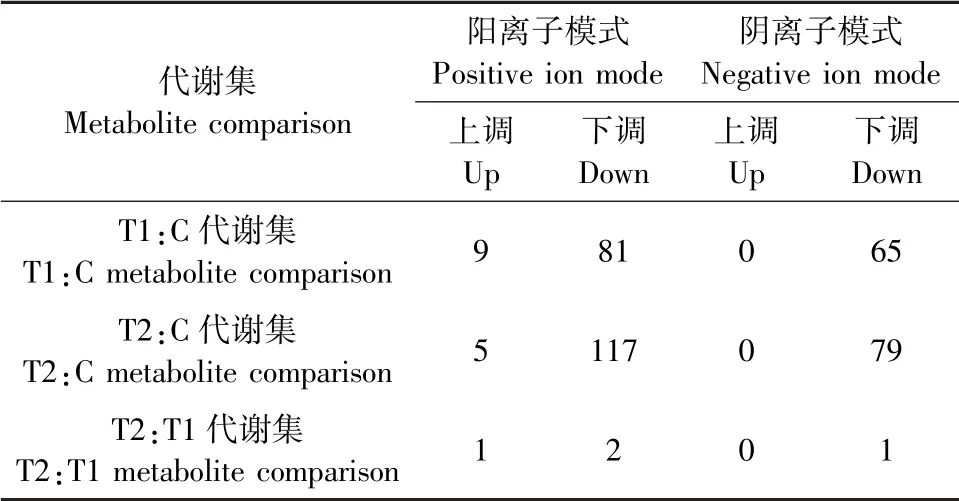
表3 差异代谢物分析结果Table 3 Results of metabolite differential analysis
由于T2:T1代谢集模型拟合不可信,因此,本研究仅对T1:C和T2:C两代谢集进行KEGG功能注释与富集分析。T2:C代谢集注释到“代谢”(metabolism)一级分类的代谢物最多。其中,“氨基酸代谢”(amino acid metabolism)二级分类通路下的代谢物个数达到为15个;其次为“脂质代谢”(lipid metabolism)有10个。类似的,T1:C代谢集注释到“代谢”一级分类下代谢物最多。其中,amino acid metabolism二级分类下代谢物为10个,和lipid metabolism分类下数目相同(图7A,7B)。此外,对代谢集内各代谢物在KEGG通路中进行通路富集和通路拓扑学分析。其中,代谢途径D-Glutamine and D-glutamate metabolism(pathway ID:map00471;P<0.01)的相对重要性最大,其次为Cutin,suberine and wax biosynthesis(pathway ID:map00073,P<0.01),它们在T1:C和T2:C代谢集中的重要性得分均高于3.0(图7C,7D)。

图7 KEGG代谢通路富集和注释结果Note.A.T1:C metabolite comparison.B.T2:C metabolite comparison.C.KEGG Topology analysis for T1:C metabolite comparison.D.KEGG Topology analysis for T2:C metabolite comparison.Figure 7 KEGG pathway classification.
2.4.4 样本中代谢物组成与微生物群落组成相关性分析
图8A,8B可知,T1:C代谢集丰度前10的差异代谢物为 2-Amino-2-methylbutanoate,(-)-Stercobilin、2,4-Dimethylpimelic acid、L-Proline、3’-Deaminofusarochromanone、 Gingerglycolipid B、Prednisolone、Spirolide D、Polysorbate 20以及PS(16∶0/18∶02(9Z,12Z)),它们与Spirochaetes的相对丰度均呈正相关。除(-)-Stercobilin外,其余差异代谢物与Spirochaetes呈显著正相关(r>0.60,P<0.05);T2:C代谢集丰度前10的差异代谢物为PE(14∶0/0∶0)、Spirolide D、Gingerglycolipid B、Prednisolone、PE(14∶1(9Z)/18∶4(6Z,9Z,12Z,15Z )、 2-Amino-2-methylbutanoate、 2,4-Dimethylpimelic acid、3’-Deaminofusarochromanone、4-[5-(3-hydroxypropyl)-1-benzofuran-2-yl]benzene-1,2-diol和Americanol,它们也与Spirochaetes的相对丰度呈正相关(r>0.60,P<0.05)。
将样品中丰度前10的菌属与上述差异代谢物进行相关性分析(图8C,8D)。结果显示,T1:C和T2:C代谢集中丰度前10的差异代谢物与Treponema_2呈正相关(r>0.6,P<0.05),而与Ruminococcaceae NK4A214 group和[Eubacterium]coprostanoligenesgroup呈负相关(r<-0.4,P<0.05)。提示粪便中这些特定菌属的丰度与差异代谢物的产生密切相关。Spirochaetes在C组平均相对丰度为8.34%。有氧暴露下FMT制备操作后丰度降低为1.28%,厌氧下丰度降低到1.13%,降低倍数超过6.5倍。Treponema_2属于Spirochaetes细菌,在C,T2和T1组中的相对丰度分别为(7.31%±4.90%),(0.69%±0.37%)和(0.81%±0.51%)。Ruminococcaceae NK4A214group属于Firmicutes细菌,在C,T1和T2组中的相对丰度分别为(2.16%±0.25%)、(3.36%±0.91%)和(3.42%±0.62%)。[Eubacterium]coprostanoligenesgroup属于Firmicutes细菌,在C、T1和T2组的相对丰度分别为(2.58%±0.37%)、(5.37%±2.33%)和(4.69%±1.01%)。这些菌群丰度受FMT制备操作而改变,并显著影响包括2-Amino-2-methylbutanoate、2,4-Dimethylpimelic acid、Prednisolone、Spirolide D等在内的多种代谢物的产生。

图8 粪便细菌和真菌群落组成与代谢物水平的相关性分析Note.A.Correlation between metabolite and phylum-level microbial composition in T1:C metabolite comparison.B.Correlation between metabolite and phylum-level microbial composition in T2:C metabolite comparison.C.Correlation between metabolite and genus-level microbial composition in T1:C metabolite comparison.D.Correlation between metabolite and genus-level microbial composition in T2:C metabolite comparison.*Significant correlation.Red color.Positive correlation.Blue color.Negative correlation.Figure 8 Correlation analysis between microbial composition and metabolites in SPF pigs’fecal samples
3 讨论
在以前的研究中,供体的作用已被证明是重要的[15]。SPF猪是在屏障或隔离环境下饲养,排除了特定的病原微生物和寄生虫的猪群,是优良的动物实验、生物制剂及生命科学研究用猪。因此,选择SPF猪的粪便作为FMT的供体具备合理性。此外,多捐赠者方法已被证实可以提高供体微生物群落多样性,有助于FMT后取得一个良好的结果[16]。因此,参照现行国家标准GB/T22914-2008,本研究最终选择了5头SPF巴马小型猪作为FMT供体动物。
供体猪粪悬液的制备方法影响了样品中菌群组成结构,这是影响FMT效果的又一重要因素。过去研究发现益生菌的活力对其发挥重用具有重要影响。然而,Andresen等[17]研究发现死菌也能对肠易激综合征发挥益处;冷冻和新鲜粪便制剂对CDI具有相似的治疗功效[18-20],提示粪便制剂中活菌数可能并不制约FMT的使用。制备FMT用的粪悬液时,短暂的有氧暴露和厌氧暴露均未显著改变样品中细菌和真菌组成,但影响了菌群的丰度。和未处理粪便样品相比,有氧和厌氧操作都完整地保留了全部的细菌门分类,猪FMT用粪悬液中仍然是以Firmicutes和Bacteroidetes为主,和健康猪群的直肠微生物研究结果一致[21]。然而,有氧和厌氧下FMT制备操作导致了Spirochaetes细菌的丰度减少约8倍,Terrisporobacter的丰度减少3倍,Treponema_2丰度减少9倍以上。此外,结果显示SPF巴马小型猪粪便中真菌群落以Ascomycota,Basidionmycota和Neocallimastigomycota为主,FMT制备操作导致了粪便中真菌群落丰度的降低。比如,有氧处理组降低了粪便中Neocallimastigomycota的丰度超过36倍,厌氧处理组降低该菌的丰度也达到20倍。值得注意的,有氧或厌氧下FMT制备操作也造成Wallemina等低丰度菌群丰度的降低,甚至无法通过扩增子测序手段检出。当然,FMT制备操作也提高了某些菌群的丰度,包括Ruminococcaceae UGC-005,Rikenellaceae RC9gutgroup以及Ruminococcaceae NK4A214 group的丰度都显著高于未处理的粪样(P<0.05)。值得注意的是,Spirochaetes含有猪肠道内与猪痢疾有关的主要病原,尤其是密螺旋体属(Treponema)细菌[22]。本研究中,5头供体猪粪便样本按照NY/T545的方法完成了猪痢疾密螺旋体病病原检测,结果为阴性。Treponema_2在15个样本中均被检出,并随FMT制备操作丰度显著降低(P<0.05)。其中,它在未处理组的平均丰度为(7.31%±4.90%),在有氧暴露组为(0.81%±0.51%),在厌氧暴露组为(0.69%±0.37%)。此外,研究显示对供体猪粪便样品实施处理后,样品中代谢物含量也发生改变,且厌氧下处理对供体猪粪便悬液中的代谢产物更具影响,毕竟与未处理组相比,厌氧暴露组中检出了201种差异代谢物,而有氧暴露组中检出155种差异代谢物。相关性分析结果揭示粪便中Treponema_2与多种代谢物的产生密切相关。值得注意的是,经FMT制备操作后Spirochaetes丰度降低,更导致了Prednisolone和Spirolide D两种代谢产物的减少。Spirolide D是一种脂溶性贝毒素,Prednisolone(中文名:波尼松龙)是常用的抗炎肾上腺皮质激素类药物,具有抗炎作用,这些结果提示本研究采用的粪便FMT制备操作方法可有效降低猪粪便中Treponema和Spirochaetes菌群的丰度,一定程度上提高了FMT的安全性。
目前,FMT不仅作为治疗复发性CDI,恢复患者肠道多样性的一种技术[23-24],还广泛用于悉生动物模型的构建,在宿主代谢、营养、免疫力以及药物发现中肠道生态学的研究上发挥着重要作用。仅从这一出发点而言,没有必要采用标准化的制备方法,但应尽可能地保证移植的粪悬液反映的是供体原始的粪便微生物组。除制备时环境空气暴露的差异外,研究使用的供体猪数量、动物个体差异以及新鲜粪便采集量、实验操作时间、以及每份测序样本中所含细菌、真菌以及代谢物含量的差异等都可能成为潜在影响因素。当然,在今后实际使用上可通过扩大供体猪数量和粪便采集量、自动化粪悬液制备、增加测序样本重复数等操作来降低这些潜在问题。
4 结论
SPF巴马小型猪粪便样品中主要的细菌群落为Firmicutes和Bacteroidetes,主要的真菌群落为Ascomycota、Basidionmycota和Neocallimastigomycota。尽管处理时间短暂,粪悬液的制备操作仍强烈地降低了包括Spirochaetes、Terrisporobacter和Treponema_2在内的细菌群落以及Neocallimastigomycota真菌群落的丰度,提高了Ascomycota的相对平均丰度,并显著影响猪粪悬液中包括Prednisolone、Spirolide D等在内的多种代谢物的产生。值得注意的是,有氧暴露对猪粪悬液中真菌群落丰度的影响高于厌氧暴露处理。结果提示,粪悬液的FMT制备操作有效地降低Treponema和Spirochaetes等有害菌群和相关代谢产物的丰度,提高了FMT的安全性。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
理化检验-化学分册(2020年5期)2020-06-15
农药科学与管理(2019年5期)2019-08-13
云南医药(2019年3期)2019-07-25
武警医学(2018年10期)2018-11-06
中成药(2017年9期)2017-12-19
当代化工研究(2016年7期)2016-03-20
质谱学报(2015年5期)2015-03-01
中国当代医药(2015年20期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
