
钠离子通道相关癫痫性脑病研究进展
2021-11-18 08:51:54袁启锋余诗倩姚宝珍
临床儿科杂志 2021年11期
袁启锋 余诗倩 姚宝珍
武汉大学人民医院儿科(湖北武汉 430060)
癫痫性脑病(epileptic encephalopathy,EE)是由反复癫痫发作或发作间期癫痫性放电所引起的疾病,频繁的癫痫活动本身导致认知和行为障碍。随着时间推移,这种损害会愈加严重,故早期有效干预可改善发育结果。3 岁内首次癫痫发作的患儿40%为EE[1],而其中30%~50%严重神经精神发育障碍患儿存在癫痫相关基因变异[2]。除频繁癫痫活动本身所造成的认知障碍和行为异常外,致病基因变异也可能造成EE 患儿神经精神发育异常,如Dravet综合征(Dravet syndrome,DS)患儿在1~2 岁时即出现神经精神发育滞后或停滞,但此时患儿脑电图上癫痫样放电并不频繁[3]。电压门控钠离子通道(voltage-gated sodium channels,VGSCs)由形成孔的α 亚基和2 个β 亚基构成,根据α 亚基不同VGSCs 分为9 种亚型(Nav1.1~Nav1.9),其中Nav1.1、Nav1.2、Nav1.3、Nav1.6 在神经元动作电位产生和传导中扮演着重要角色,其α 亚基分别由SCN 1 A、SCN 2 A、SCN 3 A、SCN8A编码,SCN1B则编码Navβ1亚基。虽已明确SCN1A、SCN2A、SCN3A、SCN8A、SCN1B变异会导致EE,但基因变异如何导致疾病的发生以及基因与临床表型的关系仍是当前亟待弄清的问题。本文综述VGSCs相关EE基因型和临床表型的关系,为EE基因诊断和精准治疗提供帮助。
1 SCN1A基因与EE
1.1 SCN1A与Nav1.1
SCN 1 A基因位于2 号染色体q 24.3,有31 个外显子,编码Nav1.1 的α亚基。Nav1.1 在兴奋性神经元和抑制性神经元上均有表达,但主要存在于表达小白蛋白的抑制性中间神经元(parvalbumin positive interneurons,PV神经元),且成年大脑表达水平更高[4]。Nav1.1 功能下调可能与多种神经精神疾病相关,如SCN 1 A基因敲除杂合子小鼠大脑PV神经元Nav1.1低表达,导致阿茨海默症发生[5]。与阿茨海默症相似,SCN 1 A杂合变异DS 模型小鼠海马CA 1 区PV神经元数量减少,且神经元上Nav1.1功能减弱[6]。提示SCN 1 A基因变异本身也可能会导致癫痫患者认知功能损害。
1.2 SCN1A相关EE
DS 是最典型的SCN 1 A相关EE,早期通常由高热诱发,随着年龄增长,部分DS 出现无热性癫痫发 作[7]。目前临床上应用较为广泛的全外显子测序技术只能发现DS患儿SCN1A外显子变异,但实际SCN1A内含子区域发生变异,诱导毒性外显子形成,进而SCN1A编码的全长蛋白数量减少,也会导致DS[8]。除此之外,某些基因可能在SCN 1 A表达的调控过程中发挥重要作用,从而影响SCN1A相关EE严重程度[9]。当然,并非所有SCN1A变异相关EE均表现为DS。有报道,9例早期婴儿SCN1A脑病中,8例为p.Thr226Met变异,1例为p.Pro1345Ser变异;9例患儿均有严重的发育障碍,无法行走和说话,甚至需通过胃造瘘方式进食[10]。p.Thr226Met位点变异时,Nav1.1离子通道功能增强,但这种功能获得会导致PV神经元更容易发展为去极化阻滞,导致PV神经元释放γ-氨基丁酸(GABA)减少,并且比DS因SCN1A变异导致单倍体剂量不足时释放的GABA更少,即前者去抑制作用更强,这就能解释早期婴儿SCN1A脑病临床表型较DS更严重[11]。见图1。由此可见,SCN 1 A外显子变异导致VGSCs功能受损程度越大,临床表型越严重,且该基因内含子变异或调控表达过程的其他基因发生变异,均可导致EE。
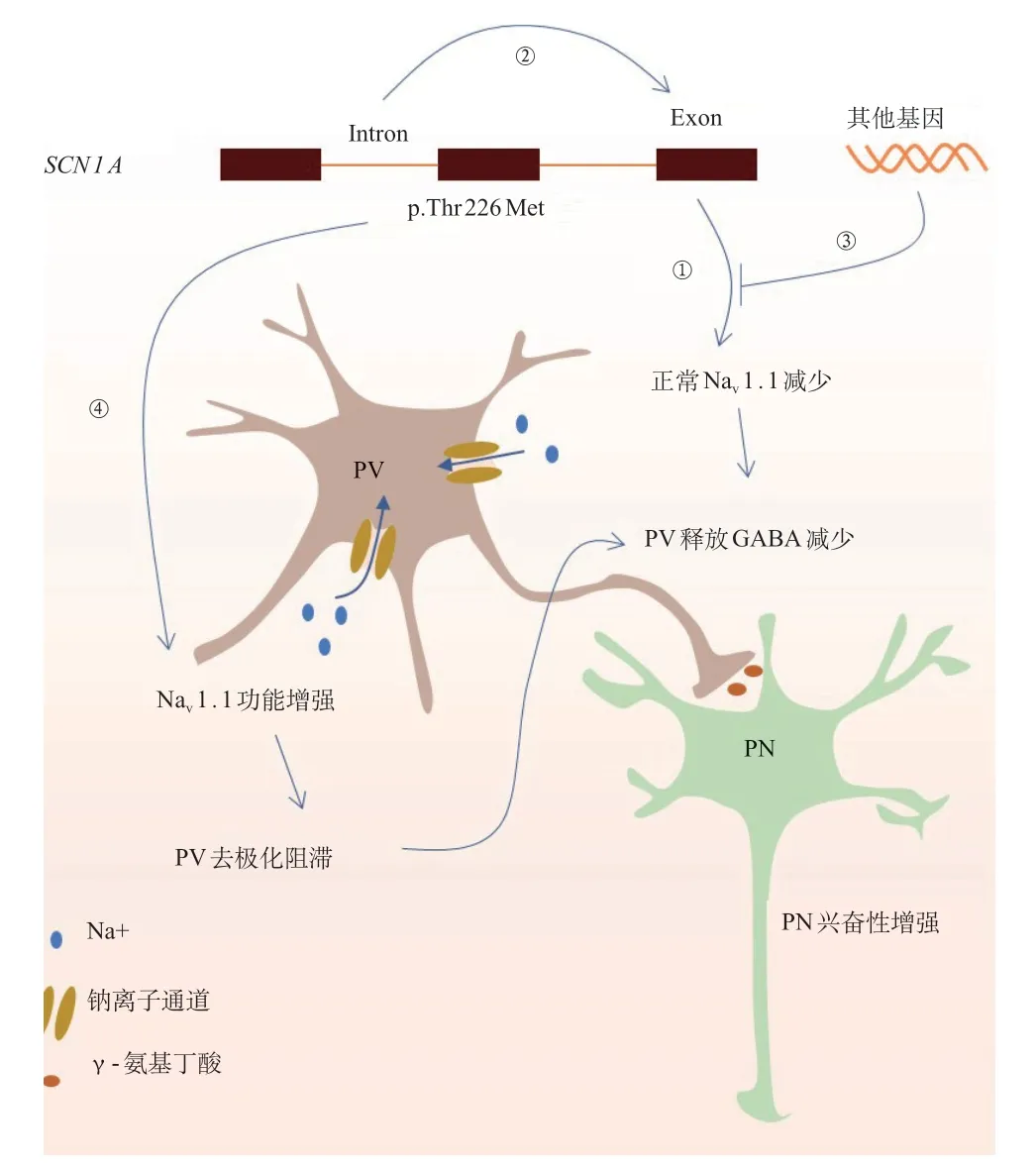
图1 SCN1A 相关EE 发病机制
1.3 SCN1A相关EE治疗
减少持续的癫痫发作和癫痫持续状态是治疗DS首先考虑的事情,因为反复癫痫发作或长时间癫痫发作会影响患儿神经精神发育,导致预后较差。从大麻中可提取大麻二酚和Δ9-四氢大麻酚,这两种药物均可有效缓解SCN1A相关DS模型小鼠癫痫发作,且两种药物小剂量静脉注射可产生协同效应[12-13]。大多数DS均由SCN1A杂合功能丧失性变异所引起,故VGSCs阻滞剂类抗癫痫药如卡马西平、拉莫三嗪、奥卡西平等无效,甚至会加重病情[14],而Nav1.1 高选择性激活剂蜘蛛毒液肽Hm 1 a 则可通过作用于大脑CA1区PV神经元,显著降低DS小鼠癫痫发作次数和死亡率[15]。由此可见,激活PV神经元抑制功能,可有效治疗DS。司替戊醇和氯巴占可增强GABA 能神经传导,控制癫痫发作。北美一项DS 治疗管理共识建议,一线抗癫痫药效果不佳时,可采用司替戊醇/丙戊酸盐/氯巴占合用方案[16],DS患儿长期维持该方案安全有效,且成年后仍可从该方案获益[14]。此外,已从SCN1A变异DS患者获取皮肤成纤维细胞,并将其成功诱导为多能干细胞[17],这将为SCN1A相关EE新药物研发及筛选带来极大可能。
2 SCN2A基因与EE
2.1 SCN2A与Nav1.2
SCN 2 A位于2 号染色体q 24.3,有33 个外显子,编码Nav1.2 的α亚基。生命早期,Nav1.2 在有髓神经纤维的轴突起始节段和Ranvier节点中表达,到1~2岁时逐渐被Nav1.6取代。成人时期,Nav1.2主要在兴奋性神经元轴突起始段及未髓鞘化轴突部位表达,此时Nav1.2 的作用是增大而非启动成熟神经元动作电位。丹麦调查发现,SCN2A基因在人群中变异率约为1/78608[18]。
2.2 SCN2A相关EE
SCN2A变异会导致多种神经精神综合征,其严重程度从自限性癫痫到伴神经精神发育障碍的EE。即使SCN 2 A不同位点发生变异,导致神经元上Nav1.2生物物理特性不同,也可能会出现相同的临床表 型[19],反之,相同位点的变异临床表型也可能不同,说明EE 的临床表型还可能受环境因素影响。有报道1例大田原综合征女性患儿及其同父异母兄弟均携带SCN 2 A上p.Q 383 E 变异,但其兄弟为良性家族性新生儿癫痫而非大田原综合征[20]。此外,变异来源也可能影响EE严重程度,胚系变异较新生变异Nav1.2电生理功能障碍更小,临床症状更轻[21]。SCN2A变异相关EE的Nav1.2电生理变化较为复杂。出生后3个月内首次癫痫发作患儿VGSCs功能增强,并且发育障碍更严重,而出生3个月后首次癫痫发作患儿VGSCs功能丧失,发育障碍相对较轻[22]。选择性剪接异变体可加重SCN2A基因变异相关EE临床症状,并且在出生前3 个月表达更高[23]。这或许能部分解释SCN 2 A基因相关EE 首次癫痫发作时间越早,患儿症状越严重的现象。
2.3 SCN2A相关EE治疗
有报道12例SCN 2 A基因变异相关EE 患儿使用苯妥英后,症状得到有效控制;但纳入更多研究对象后发现,出生3 个月内首次癫痫发作脑病患儿在使用VGSCs 阻滞剂(苯妥英、卡马西平)后癫痫发作频率明显减少甚至完全控制;相反,出生3 个月后首次癫痫发作脑病患儿使用VGSCs阻滞剂,癫痫控制效果甚微,甚至会加剧癫痫发作[24]。这与SCN2A相关EE发病机制相一致,也说明研究VGSCs功能变化对于该类疾病的治疗至关重要。此外,部分SCN 2 A变异EE 患儿的父亲或母亲为嵌合体变异,其下一个孩子可能有患EE的风险,故备孕前遗传咨询对预防EE的发生是必要的[25-26]。
3 SCN8A与EE
3.1 SCN8A与Nav1.6
SCN 8 A位于12 号染色体q 13.13,有28 个外显子,编码Nav1.6的α亚基。微管相关蛋白MAP1B与Nav1.6 胞内N 末端结合,使得Nav1.6 不被内吞并在神经元轴突起始段稳定存在[27]。磷酸化是一种强大的可逆的离子通道功能调节机制,钙离子/钙调蛋白依赖性蛋白激酶II可通过调节Nav1.6磷酸化,进而影响神经元兴奋性[28]。同样,Janus激酶2通过使细胞内成纤维细胞因子14磷酸化,调节Nav1.6功能,从而抑制Janus激酶2活性,导致海马CA1区锥体神经元兴奋性降低[29]。可见,磷酸化可调节Nav1.6功能,而Nav1.6功能变化则会影响锥体神经元兴奋性。
3.2 SCN8A变异相关EE
SCN8A相关EE一般是由2个杂合变异亲代遗传所致,子代为双等位基因变异,并且这2个变异都表现为VGSCs 活性降低[30]。严重SCN 8 A相关EE 的特征是在出生后最初几个月出现顽固性癫痫,通常表现为长时间的局灶性运动减退,并成串发作,伴有明显的植物神经症状(呼吸暂停、发绀、瞳孔扩大),演变为阵挛或双侧强直-阵挛表现[31]。内侧内嗅皮层接收顶叶、枕叶所传入的信息,并将其传出到海马锥体神经元,SCN8A基因变异导致该区域神经元Nav1.6钠离子通道失活障碍、钠离子电流增大[32]。巧合地是,SCN8A基因变异相关EE小鼠海马CA1区锥体神经元也会出现过度兴奋[33],这两种现象之间或许存在因果关系,并可解释SCN 8 A基因变异相关EE 的发病机制,即SCN8A基因变异导致内侧内嗅皮层神经元Nav1.6钠离子通道失活障碍,进而海马CA1区神经元过度兴奋,EE发生。此外,前脑(大脑皮层和间脑)兴奋性神经元Nav1.6钠离子通道失活障碍也会导致SCN8A基因变异相关EE发生[34]。由此可见,神经元Nav1.6钠离子通道失活障碍在SCN8A基因变异相关EE发生过程中扮演重要角色。然而,EE的发生发展并非仅由Nav1.6功能变化所影响。癫痫发作后,一些其他基因在前脑表达水平的继发性变化也可能会导致脑病进展[35]。
3.3 SCN8A变异相关EE治疗
VGSCs 阻滞剂对SCN 8 A相关EE 发作有较好的控制作用,尤其是高选择性拮抗Nav1.6和Nav1.2、保留Nav1.1功能且容易透过血脑屏障的钠离子拮抗剂,对SCN8A基因相关EE的治疗具有重大意义。GS967是一种新型VGSCs 阻滞剂,高选择性减少钠离子电流。研究发现,GS 967 抑制锥体神经元自发放电,降低海马Nav1.6蛋白水平,但不影响PV神经元动作电位放电频率,可有效缓解SCN 8 A相关EE 症状[36-37]。Prax330是另一种新型钠离子拮抗剂,可减小VGSCs持续电流和复活电流,抑制动作电位爆发,并降低下丘脑神经元兴奋性,对SCN 8 A相关EE 也有较好的疗效[38]。此外,基因治疗在将来也会是一个很好的选择,如反义寡核苷酸将SCN 8 A基因转录丰度降低25%~50%,可有效减少EE 小鼠癫痫发作次数,延长小鼠生存时间[39]。
4 SCN3A与EE
由SCN3A编码的Nav1.3主要在儿童发育过程中表达。发育阶段,SCN 3 A富集在基底/外向径的神经胶质细胞群和正在迁徙的新生神经元群中。SCN3A变异可能导致神经元异位,MRI 可发现大脑结构缺陷,如外侧裂周边区多微脑回或双侧额顶叶多微脑回伴小脑畸形[40],且患儿表现为神经精神发育异常[41]。
首例报道SCN 3 A基因变异相关EE 患儿在出生后几周内即表现为顽固性癫痫和严重的智力残疾,VGSCs 功能分析显示明显的功能获得性效应,缓慢失活电流显著增加;苯妥英和拉科酰胺选择性减少缓慢失活电流,能有效缓解患儿的临床症状[42]。另1例SCN3A基因相关EE患儿纯外显子测序未发现基因变异,但外显子组隐马尔可夫模型发现患儿存在SCN3A基因位点缺失[43]。随着基因检测技术的发展,未来可能发现更多EE与SCN3A变异相关。
5 SCN1B与EE
Navβ 1 亚基在神经系统广泛表达,由SCN 1 B基因编码,与VGSCs 在细胞表面定位密切相关。该基因杂合变异可表现为热性惊厥附加症等非脑病性癫 痫[44],纯合隐形变异则表现为DS 样EE 症状,且这类患儿生命早期死亡率较高[45-46]。与典型DS 相比,SCN 1 B基因纯合变异患儿临床症状更符合早期婴儿发育和癫痫性脑病(developmental and epileptic encephalopathy,DEE),后者比DS更严重[47]。与野生型细胞相比,SCN1B基因变异(p.Arg85Cys)细胞表面钠离子通道β 1 亚基表达并无差异,但由β 1 亚基调控的Nav1.1钠离子电流消失,表明SCN1B该位点变异是功能丧失性的[44]。高选择性激活Nav1.1 或许可有效缓解该类EE症状。Na+-K+-2 Cl-同向转运体1型(Na+-K+-2Cl-co-transporter-1,NKCC1)和K+-Cl-同向转运体2 型(K+-Cl-co-transporter-2,KCC 2)是锥体神经元上2 种主要的Cl-通道,前者介导Cl-内流,后者介导Cl-外流,在大脑发育早期,细胞表面NKCC1表达为主,神经元内Cl-浓度高,当GABA受体被激活,胞内Cl-外流,神经元去极化,随着神经元成熟,细胞表面NKCC1表达逐渐减少,而KCC2表达逐渐增多,神经元外Cl-浓度高,此时激活GABA受体则表现为神经元超极化。研究发现,SCN1B变异相关EE小鼠的大脑GABA能抑制作用成熟延迟,神经元过度兴奋,导致癫痫发生[48](图2)。研发NKCC1抑制剂有望治疗SCN1B相关EE。
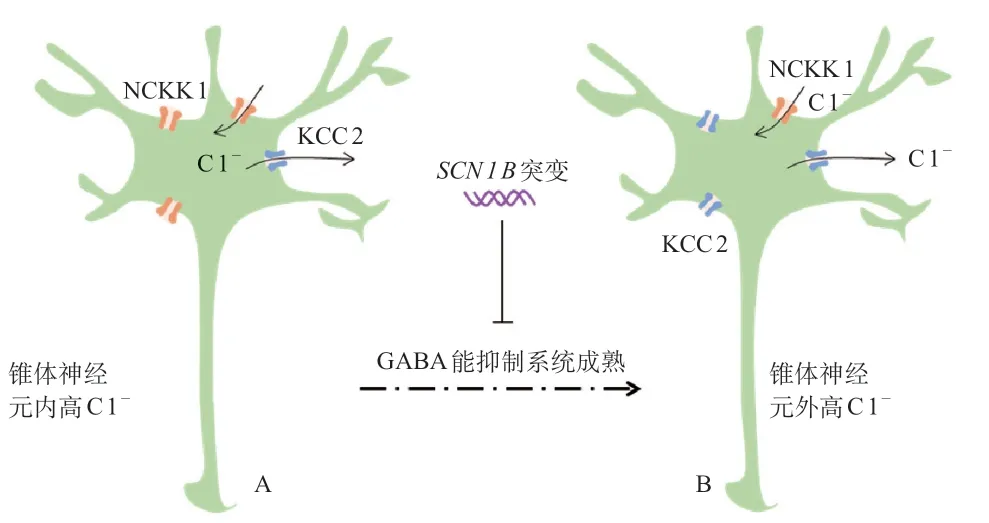
图2 SCN1B 变异EE 小鼠GABA 能抑制系统成熟延迟示意图
综上,理解VGSCs基因变异和EE 的生物学概念有助于阐明其基因异质性和表型异质性,并为临床实践提供指导。如果能根据临床表型明确致病基因,则进行特异性基因测序将有助于诊断。但是,由于同一基因发生变异可能表现为不同的临床特征,相同的临床表型也可能由不同基因变异所致,那么,从遗传学角度定义描述EE 将是一种趋势。随着基因诊断在临床上逐渐推广,其指导理解脑病发病机制和治疗策略的潜力不可忽视。在VGSCs 相关EE 的治疗中,根据基因型对治疗分类,将使之朝着精准医疗的方向迈进。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
仪器仪表用户(2021年10期)2021-11-27 08:26:14
中国生殖健康(2020年4期)2021-01-18 02:58:10
世界最新医学信息文摘(2020年68期)2020-12-25 11:55:27
中国生殖健康(2018年4期)2018-11-06 07:12:16
中华老年多器官疾病杂志(2016年2期)2016-01-16 03:15:46
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:13
电源技术(2015年2期)2015-08-22 11:28:30
物理化学学报(2015年5期)2015-02-28 17:34:57
湖北农业科学(2014年11期)2014-09-10 18:06:07
