
长沙市新生儿听力和聋病易感基因联合筛查的临床分析
2021-11-13 00:43秦华丽蔡岳祥欧阳耿
中国耳鼻咽喉颅底外科杂志 2021年5期
秦华丽,蔡岳祥,欧阳耿
(1.长沙市妇幼保健院 眼耳鼻咽喉科,湖南 长沙 410007; 2.南华大学附属长沙市中心医院 院办公室,湖南 长沙 410018)
2006年全国第二次残疾人抽样调查结果发现,我国听力残疾人数高达2 780万,未残疾注册而存在的听障人数远多于此。小于7岁的达80万,每年新增6~8万,数量巨大[1]。因此,耳聋的防治一直受到我国政府及各级主管部门的高度重视。我国已经形成了非常成熟完善的新生儿听力筛查流程[2],但对潜在的耳聋高危儿及迟发性聋存在着些局限性[3],据已知的流行病学研究显示,大约60%的耳聋患者与遗传因素有关,还有40%为环境或未知因素。根据全国聋病分子调查,在遗传性耳聋中GJB2,SLC26A4,MTRNR1和GJB3这4个最高发的基因,各自有着最高发的突变热点[4],随着聋病基因诊断技术的开展应用,2007年提出了听力和聋病易感基因联合筛查的理念[5],为优化聋病防控提供了新的模式。我国诸多地区对新生儿进行了听力和耳聋基因联合筛查,北京[6]、天津[7]、广东[8]、广西[9]、云南[10]等地分析了新生儿听力和耳聋基因联合筛查结果,目前尚缺乏长沙市新生儿的联合筛查大数据,本研究对长沙市5 526例新生儿听力筛查和聋病易感基因检测结果分析,为全国的联合筛查提供新的支持数据。
1 对象与方法
1.1 研究对象
选择2017年5—12月在长沙市出生的新生儿为研究对象,出生3~7 d采集足跟血检测耳聋基因。所有参与新生儿耳聋基因筛查的新生儿家长均认真阅读并签署知情同意书。共收集样本5 526例,其中3 708例母婴同室新生儿,1 818例住进新生儿科。
1.2 听力筛查方法
采用 Accusceen 听力筛查仪(丹麦产)进行新生儿耳声发射(otoacoustic emission,OAE)检查。接受初筛的年龄为出生后 2~3 d(48~72 h),出生后转新生儿科住院的患儿出院前完成筛查。初筛显示未通过的新生儿,出生后 42 d(早产儿则在修正月的36~44 d)复查OAE并进行快速听性脑干反应(rapid auditory brainstem response,AABR)检查。以上测试环境及操作要求:噪声控制于45~50 dB(A)以下,新生儿耳道尽量保持干燥干净无异物,选择与新生儿耳道大小合适且清洁的耳塞,在安静、睡眠状态下进行操作。AABR 以Ⅴ波反应阈(正常听力级)<35 dB 为通过。初筛、复筛均未通过的婴幼儿满3个月时做诊断性听力学检查。
听力诊断,以听性脑干反应(auditory brainstem response,ABR)反应阈作为高频听力损失的参考指标:轻度为31~50 dBnHL,中度为51~70 dBnHL,重度为71~90 dBnHL,极重度为>90 dBnHL.
1.3 聋病易感基因筛查
出生3~7 d采集足跟血斑,共2枚,用于筛查和验证。所有参与新生儿耳聋基因筛查的新生儿家长均认真阅读并签署知情同意书,共收集样本5 526例,应用北京博奥晶典生物公司的9项遗传性耳聋基因检测试剂盒,试剂盒采用多重等位基因特异性PCR结合通用芯片技术对检测4种耳聋常见遗传基因9个位点,包括GJB2基因(35delG、176-191del16、235delC、299-300del AT4);SLC26A4基因(IVS7-2A>G、2168 A>G);MTRNR1基因(1555A>G、1494C>T);GIB3基因538C>T。
1.4 随访
听力筛查结果当面告知家长,耳聋基因筛查结果以电话及短信方式告知家长,不能回院检查者电话随访,对未通过的新生儿重点随访,定期随访其听力、言语发育及干预情况和防聋指导。
1.5 统计学方法
应用SPSS 25.0软件,用频数、百分比和χ2检验进行数据统计分析,组间χ2检验采用双侧检验,以P<0.05表示差异具有统计学意义。
2 结果
2.1 听力筛查情况
5 526例中,初筛未通过667例(12.07%,667/5 526),其中631例(94.60%,631/667)接受复筛, 结果107例未通过,复筛未通过率为16.95%(107/631),复筛未通过的107例在3月龄时经进行了听力学诊断,最终确诊听力损失患儿14例(23耳),其中双耳极重度3例,重度2例,中度2例,轻度1例,单耳听力损失极重度3例,中度1例,轻度1例,还有1例左耳极重度右耳轻度听力损失。其中7例(10耳)听力异常者,耳聋基因筛查正常。随访8耳佩戴助听器,4 耳人工耳蜗植入。见表1。
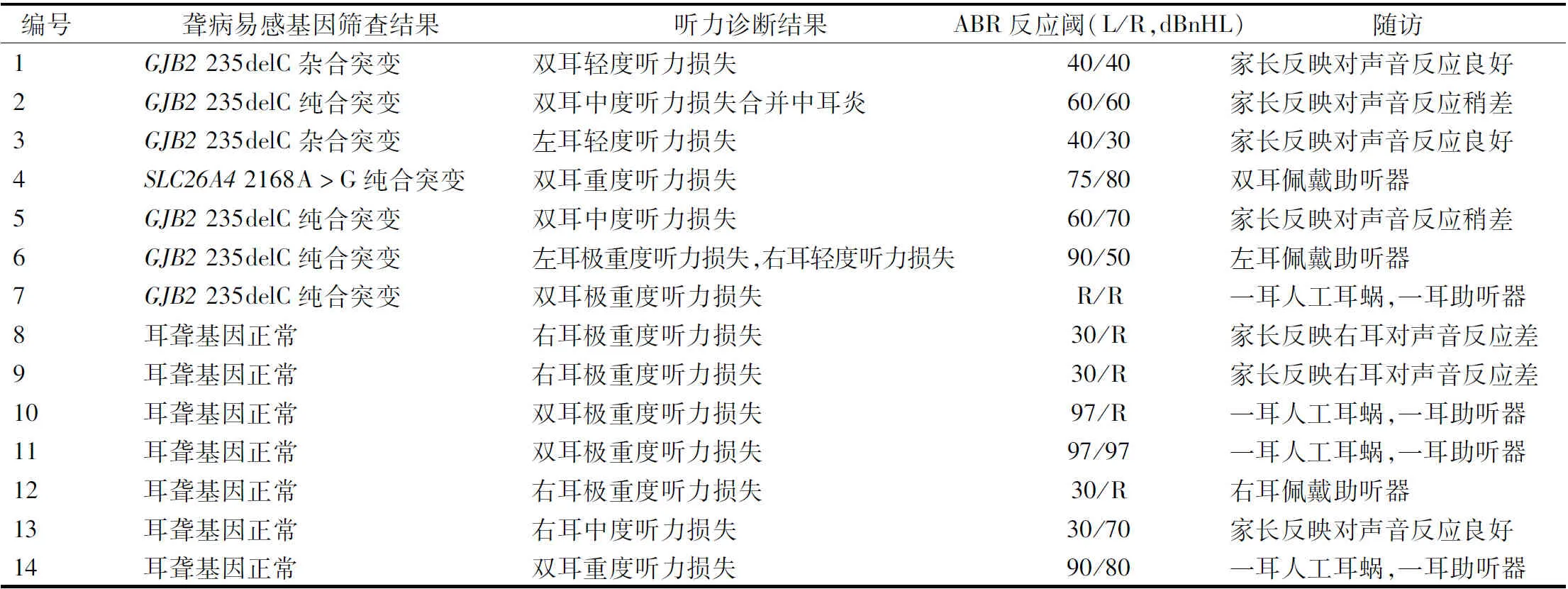
表1 14例听力损失者聋病易感基因筛查和听力诊断
2.2 聋病易感基因筛查情况
5 526例中,筛查未通过234例,其中纯合突变5例,包括GJB2235delC纯合突变4例,SLC26A42168A>G纯合突变1例;单位点杂合突变229例(累计多位点杂合突变计数为233例),包括GJB2235delC杂合/MTRNR11555均质1例,GJB2235delC/SLC26A42168A>G双杂合1例,GJB2235delC/SLC26A4IVS 7-2A>G双杂合突变2例。GJB2235delC杂合突变118例,176-191del16单杂合突变5例,299-300 del AT单杂合突变14例,SLC26A4单杂合突变66例,MTRNR1基因突变20例,GJB3538C>T单杂合突变6例。见表2。

表2 5 526例新生儿聋病易感基因突变情况 [ 例(%)]
2.3 听力和聋病易感基因联合筛查情况
5 526例受检者中听力初筛和基因筛查均通过4 728例,听力初筛和基因筛查均未通过103例,听力初筛通过4 859例中,耳聋基因筛查未通过131例,未通过率为2.69%(131/4 859);听力初筛未通过的667例中,耳聋基因筛查未通过103例,未通过率15.44%(103/667),经χ2检验,两者差异具有统计学意义(χ2=234.97,P=0.000)。见表3。
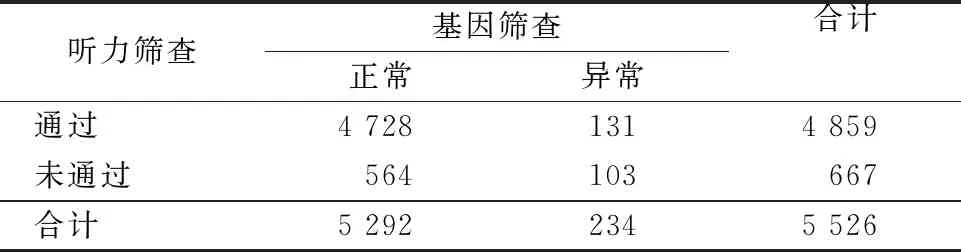
表3 5 526例听力初筛与聋病易感基因筛查结果 (例)
3 讨论
本研究分析显示,听力初筛未通过的新生儿中聋病易感基因筛查未通过率15.44%(103/667)高于听力筛查通过者 2.69%(131/4 859),且差异有统计学意义(P<0.000),验证了聋病易感基因异常与听力筛查未通过的相关性,提示联合筛查未通过的互为随访的高危人群。本研究中4个聋病易感基因的异常率4.23%(234/5 526),后者比Chen等[11]分析的异常率4.38%稍低,可能与样本量少有关。从研究结果来看,5.40%(36/667)听力初筛未通过的父母自觉孩子对声音反应良好,还有耳聋基因筛查异常不愿随访的,要利于家长的理解接受及随访工作的开展,联合筛查结果必须由遗传优生科与耳鼻喉科等多专业学科共同解读。
GJB2基因突变有地域性,在亚洲该基因最常见的突变位点是235delC[12]。GJB2基因突变常见于双耳感音神经性耳聋,表型多样,与GJB2基因突变的类型和位点有关[13-14]。本研究中,GJB2基因杂合突变率2.48%(137/5 526),与2019年北京[15]新生儿GJB2基因突变率2.419%稍高;4例GJB2 235delC纯合突变全部出现不同程度的听力损失,2例为双耳中度听力损失,1例双耳极重度听力损失,1例左耳极重度,右耳轻度听力损失,其听力表型多样化,与多项研究结果一致[13];其中1例GJB2 235delC纯合突变患儿,双耳极重度听力损失,6月龄时双耳佩戴助听器,1岁时,1耳植入人工耳蜗,1耳佩戴助听器,目前言语发育尚可,与多项研究表明GJB2基因导致的极重度感音神经性耳聋患儿接受人工耳蜗植入后听力语言康复效果好的结果一致[16]。还有1例GJB2 235delC纯合突变患儿,左耳极重度听力损失右耳轻度听力损失,左耳佩戴助听器,目前言语发育尚可。可见通过听力学检查评估了听功能,结合耳聋基因筛查明确了病因,有利于精准治疗,避免聋哑。另外,本研究中的78例GJB2 杂合突变通过了听力初筛,听力复筛未通过的,3月龄听力诊断发现了1例双耳轻度听力损失,1例左耳轻度听力损失,这与研究者发现大约3.8%~6.9%的GJB2基因突变儿童能够通过新生儿听力筛查,但是会发生迟发性听力损失的结果是相符的[17-18],本研究概率略低(2.56%,2/78),可能与本研究样本量少有关,同时也很好地说明了耳聋基因筛查可以及早发现迟发性耳聋,是对听力筛查的有效补充。贺骏等[19]对GJB2或SLC26A4基因单杂合变异进行测序,发现同时携带其他突变位点的概率较高,强烈建议完善GJB2基因的Sanger全测序。
有研究表明,大前庭导水管综合征起病隐匿,33.15%的患儿至少有一耳通过了新生儿听力筛查[20],多数在出生后1~3岁内发病[21]。本研究中发现SLC26A4 2168A>G纯合突变1例,该患儿出生时听力筛查双耳未通过,3月龄听力学诊断为双耳重度听力损失(左耳75 dBnHL,右耳80 dBnHL),行影像学提示大前庭导水管扩张,随访6月龄时双耳佩戴助听器,摔跤或感冒后出现听力损失加重(左耳90 dBnHL,右耳95 dBnHL),及时就医治疗后又恢复到以前的听力水平,提示耳聋基因筛查可较早发现耳聋的病因,早期发现听力波动性下降,及时干预,可使大前庭导水管综合征患者的听力维持在一定的水平或使之听力下降的速度明显减缓,这与此前大部分研究结果相符[21-22];肖彩霞等[23]研究发现SLC26A4杂合突变者也有可能出现耳聋,于晓宇等[24]研究发现非综合征性大前庭导水管耳聋患者有相当一部分患者未检出SLC26A4双等位基因突变,建议本研究发现的66例SLC26A4 杂合突变者完善SLC26A4基因全序列检测,并告知家长定期听力检测及保护听力的注意事项,避免使颅内压增高的诱因,保存听力,提高生存质量及获得较多的教育学习机会。
MTRNR1基因由于核修饰基因的存在[25]、遗传异质性、阈值效应等线粒体“遗传瓶颈效应”,导致该基因异常者接触或未接触耳毒性药物听力表型差异较大[26],刘日渊等[27]研究发现MTRNR1基因阳性家系中,存在对氨基糖苷类药物不敏感的个体。本研究中母系遗传MTRNR1基因阳性检出率0.36%(20/5 526),比2019年北京[15]的0.18%高,随访中发现1例1555A>G均质突变者,其母亲服用过氨基糖苷类药物,但是目前听力及言语交流无异常,与以往研究结果一致[27]。亦有观点认为[28]m.A1555G可能存在低异质率的突变,故下一代测序的发展有可能检测出更多的假阴性个体,提高检测灵敏度。随访发放患儿听能管理注意事项卡片,谨防出现“一针致聋”。本研究中,GJB3 538 C>T单杂合突变6例(0.11%,6/5 526),比东莞的0.14%稍低[29],目前听力均通过,定期检测听力,提防迟发性耳聋。
在本研究中确诊的7例耳聋基因正常但听力损失的患儿,均有新生儿科住院史,有早产、低体重、重度高胆红素血症、病毒感染等与听力损失有关的高危因素,说明有些听力损失可能是非遗传因素引起的[30],目前基因热点筛查不能发现所有的新生儿听力损失[12]。高胜利等[31]研究发现儿童单侧重度及极重度感音神经性聋患有内耳结构异常比例较高,本研究随访发现2例右耳极重度耳聋,还有1例右耳中度耳聋,患儿家长认为左耳听力正常,能听会说,不愿采取助听干预及影像学检查,建议完善家庭成员全外显子测序有助于明确造成听力损失的病因,注重单侧耳聋的中枢重塑[32],达到双耳聆听的效果。所以听力筛查和耳聋基因筛查是不可互相取代,是互相弥补的,单一的检查结果是不能给家长全面准确的信息告知孩子的听力状况和预后的,两者结合具有早期发现听障儿童和潜在听障高危儿的优势和重要性,便于精准治疗的开展。
本研究的一些不足之处:此次研究样本量较少,研究对象来自长沙市妇幼保健院出生的新生儿,未能全覆盖长沙市全体人群样本,对研究基因只做了一代测序,但本研究结果仍可作为长沙地区耳聋基因型和相关听力表型的一个参照。
综上所述,聋病易感基因和听力筛查未通过的互为随访的高危人群,新生儿听力和聋病易感基因联合筛查可行性强,相互裨益,是目前最佳的防聋筛查模式。
猜你喜欢
昆明医科大学学报(2021年5期)2021-07-22
中国民间疗法(2021年8期)2021-07-22
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
中华养生保健(2020年3期)2020-11-16
World Journal of Clinical Cases(2019年6期)2019-04-17
中国生殖健康(2018年4期)2018-11-06
外语教学理论与实践(2016年1期)2016-06-11
中国卫生标准管理(2015年3期)2016-01-14
中国中医药现代远程教育(2014年13期)2014-03-01
