
Responsible genes in children with primary vesicoureteral reflux:findings from the Chinese Children Genetic Kidney Disease Database
2021-11-06 01:00:32JiaLuLiuQianShenMingYanWuGuangHuaZhuYuFengLiXiaoWenWangXiaoShanTangYunLiBiYiNvGongJingChenXiaoYanFangYiHuiZhaiBingBingWuGuoMinLiYuBoSunXiaoJieGaoCuiHuaLiuXiaoYunJiangShengHaoYuLinKangYingLiangGong
World Journal of Pediatrics 2021年4期
Jia-Lu Liu · Qian Shen · Ming-Yan Wu · Guang-Hua Zhu · Yu-Feng Li · Xiao-Wen Wang · Xiao-Shan Tang ·Yun-Li Bi · Yi-Nv Gong · Jing Chen · Xiao-Yan Fang · Yi-Hui Zhai · Bing-Bing Wu · Guo-Min Li ·Yu-Bo Sun · Xiao-Jie Gao · Cui-Hua Liu · Xiao-Yun Jiang · Sheng Hao · Yu-Lin Kang · Ying-Liang Gong ·Li-Ping Rong · Di Li · Si Wang · Duan Ma · Jia Rao · Hong Xu · for Chinese Children Genetic Kidney Disease Database (CCGKDD), “Internet Plus” Nephrology Alliance of the National Center for Children’s Care
Abstract
Keywords Children · Congenital anomalies of the kidney and urinary tract · Gene mutation · Vesicoureteral reflux · Wholeexome sequencing
Introduction
Primary vesicoureteral reflux (VUR) is the abnormal retrograde flow of urine from the bladder into the ureters/kidneys. It is associated with an increased risk of urinary tract infection (UTI), renal scarring, and consequent proteinuria and hypertension [ 1].
VUR is a mild congenital anomaly of the kidney and urinary tract (CAKUT) phenotype. CAKUTs are phenotypically variable and may affect the kidney(s) and/or the urinary tract [ 2- 4]. CAKUTs are the most common causes of end-stage renal disease (ESRD) in children. Other forms of CAKUT, such as renal hypoplasia/dysplasia, commonly occur with VUR. CAKUT may occur as an isolated condition or with extrarenal manifestations as part of a syndromic disorder [ 2- 4].
VUR/CAKUT results from faulty urinary system development, which relies on a process of critical multiple gene networks [ 5]. Polycystic kidney disease and ciliopathies have relatively normal early development, but cysts develop later,and there is a secondary, induced nephron defect. Therefore,we do not consider these conditions as CAKUT [ 6].
A genetic diagnosis was confirmed in approximately 1.3-21% of CAKUT patients [ 7- 16]. A variety of inheritance patterns have been observed, including autosomaldominant with incomplete penetrance, autosomal recessive,X-linked, and polygenic [ 17]. Most studies only included severe CAKUT phenotypes. No large-scale study described the genetic responsibility in children with primary VUR,especially isolated VUR. The present study identified the possible genetic contributions in these patients.
Methods
Children with primary VUR were enrolled and analysed based on a national multi-center registration network (Chinese Children Genetic Kidney Disease Database, CCGKDD,www. ccgkdd.com.cn) that covered 23 different provinces/regions in China from 2014 to 2019. 67.5% (n= 256) of the cases were from the Eastern China, 25.6% (n= 97) from the Central China, 5.8% (n= 22) were from the Western China,0.3% (n= 1) was from foreign country, and the origin of the rest 0.8% (n= 3) of cases were unknown.
Patients with symptoms suspected with VUR, such as acute pyelonephritis, a history of recurrent UTIs, or chronic kidney disease of unknown etiology, received voiding cystourethrogram (VCUG) in the medical centers. VUR was graded using the International Reflux Study Committee classification system [ 18]. In cases with bilateral VUR, the higher grade was recorded. Syndromic VUR was defined as VUR with extrarenal complications, such as ocular abnormalities and cardiovascular disease. Following data were collected from patient medical records: diagnosis of the proband, parental consanguinity, and family history related to VUR. Genetic causes of patient with diagnosed VUR were sought using whole-exome sequencing (WES) or targeted-exome sequencing.
Each medical center identified all eligible patients after standardized training from the “Internet Plus” Nephrology Alliance of National Center for Children’s Care.
Genetic tests were performed using Wuxi NextCODE,Chigene, and MyGenostics. Genomic DNA was isolated from blood lymphocytes. Exome capture was performed using Agilent SureSelect human exome capture arrays(V5, Life Technologies), NimbleGen, the xGen Exome Research Panel v1.0 (IDT) or MyGenostics Gencap capture technology followed by next-generation sequencing on the Illumina HiSeq sequencing platform. Sequence reads were mapped to the human reference genome assembly (NCBI build 37/hg19). Variant interpretation was done by a panel of nephrologists or molecular geneticists with domain expertise in inherited kidney diseases, bioinformaticians and clinical molecular geneticists performed variant interpretations using the American College of Medical Genetics (ACMG)guidelines [ 19]. Decision-making of identified variants was performed using a previously reported strategy [ 8].The databases for minor allele frequency (MAF) annotation included genomAD, dbSNP (Single Nucleotide Polymorphisms), Exome Aggregation Consortium (ExAC) and the 1000 Genomes Project. Provean, Sift, Polypen2_hdiv,Polypen2_hvar, Mutationtaster, M-Cap, and Revel software packages were used to predict protein product structure variation. Variants classified as pathogenic or likely pathogenic or of uncertain significance were confirmed using Sanger sequencing.
The Institutional Review Board (IRB) of the Children’s Hospital of Fudan University approved and monitored this study, which involved seven participating centers (no.2018286).
Statistical analysis
The patients were divided into two groups according to the genetic results. The Chi-square test or Fisher’s exact test was used to compare proportions. Kaplan-Meier survival curve was used to assess renal survival. The top four mutant genes were selected and Cox regression method was conducted to identify the prognostic mutant signature. All analyses were performed using SPSS version 19 statistical software in default settings.P< 0.05 was set as the level of statistical significance.
Results
Genetic sequencing and analysis workflow are shown in Fig. 1. A total of 379 unrelated patients (male:female,219:160) with primary VUR were recruited. A monogenic cause was identified in 28 patients (7.39%) (Table 1). The corresponding proportions in male and female patients were 7.3% (16 of 219) and 7.5% (12 of 160), respectively.These genes includedPAX2(n= 4),TNXB(n= 3),GATA3(n= 3),SLIT2(n= 3),ROBO2(n= 2),TBX18(n= 2), and the other 11 genes (one gene for each patient). These genes have autosomal-dominant inheritance (AD), exceptITGA8,CENPFandNPHP1, which exhibit autosomal recessive inheritance (AR). Additionally, 28.6% (8 of 28) of the mutations in patients with primary VUR were truncation mutations, including nonsense and frameshift mutations.

Fig. 1 Genetic sequencing and analysis workflow
Segregation analysis was performed in 17.9% (5/28) of the monogenic VUR patients. Sanger sequencing validated two possible disease-causing heterozygous dominant DNA variants inPAX2andUMODin two of our 28 studied families (Table 1). The variants in these genes showed segregations that were consistent with dominant autosomal inheritance because they were seen only in affected individuals.
One patient with unilateral high-grade reflux and contralateral renal dysplasia carried compound heterozygous mutations inCENPF.This patient also had congenital hyperopia and microcephaly. Segregation analysis revealed that each of the parents was heterozygous for one of the two variants.
Two variants that did not show consistent co-occurrence with a VUR phenotype included two known VUR genes,HNF1B(p.Ala122Ser) andPAX2(p.Y73X,324), which were inherited from healthy fathers in the respective family. The variant inHNF1Bwas a predicted deleterious, missense variant.
One patient with unilateral low-grade reflux and bilateral small kidneys, which were thought to represent renal hypo-/dysplasia, actually represented small cystic kidneys due to a homozygous deletion of theNPHP1gene, which was confirmed using real-time polymerase chain reaction.One patient with bilateral reflux and small left kidney carried a variant inUMOD(p.Arg588Stop, 53). These results changed the diagnoses.
Overall, 12.7% (48 of 379) of the children had extrarenal manifestations. The most common coexisting anomalies were congenital heart defects, which accounted for 37.5% (18/48) of all syndromic cases. This anomaly was followed by other anomalies, such as osseous anomalies (13 cases), mental retardation (13 cases), and ocular anomalies(8 cases). Patients with syndromic VUR had a higher rate of gene mutations (14.1%) than patients without extrarenal complications (6%;P= 0.035, Fisher’s exact test) (Table 2).Patients withPAX2variants had syndromic features more frequently (75.0%) than those without variants (16.3%;P= 0.016; Table 2).
The frequency of genetic abnormality was not statistically significant based on the coexistence of other CAKUT(9.6% vs. 5.6%,P= 0.139, Chi-square test) (Table 2). Mutations in theRET,TNXB,COL4A1,SLIT2,GATA3,ITGA8,
ROBO2, andTBX18genes were identified as causative in non-syndromic isolated VUR.
Children were divided into those with nondilated (grades I and II) and dilated (grades III and IV and V) VUR. We found no significant difference in the mutation rates: 9.4%vs. 6.7%,P= 0.429. Eighty-eight patient (23.2%) exhibited impaired renal function (defined as estimated glomerular filtration rate < 90 mL/min/1.73 m 2 ) at their last visit, and 18 of them (20.5%) developed ESRD at the median age of 7.0(IQR 0.9-11.4) years. Kaplan-Meier survival curve showed that the presence of genetic mutations did affect renal survival (Log-rank test,P= 0.01; Fig. 2 a). Renal survival for each gene mutation group is shown in Fig. 2 b.PAX2mutation carriers (HR 5.1, 95% CI 1.3-20.0;P= 0.02), andTNXBmutation carriers (HR 20.3, 95% CI 2.4-168.7;P= 0.01)were associated with increased risk of progression to ESRD(Table 3).
Discussion
This report is the largest known study to identify genetic factors of primary VUR. A monogenic mutation was discovered in 7.3% cases, which is similar to previous studies using various detection methods [ 7- 16].
Many genes have been reported to cause VUR among unrelated patients, such asPAX2,RET,ROBO2,TNXB,
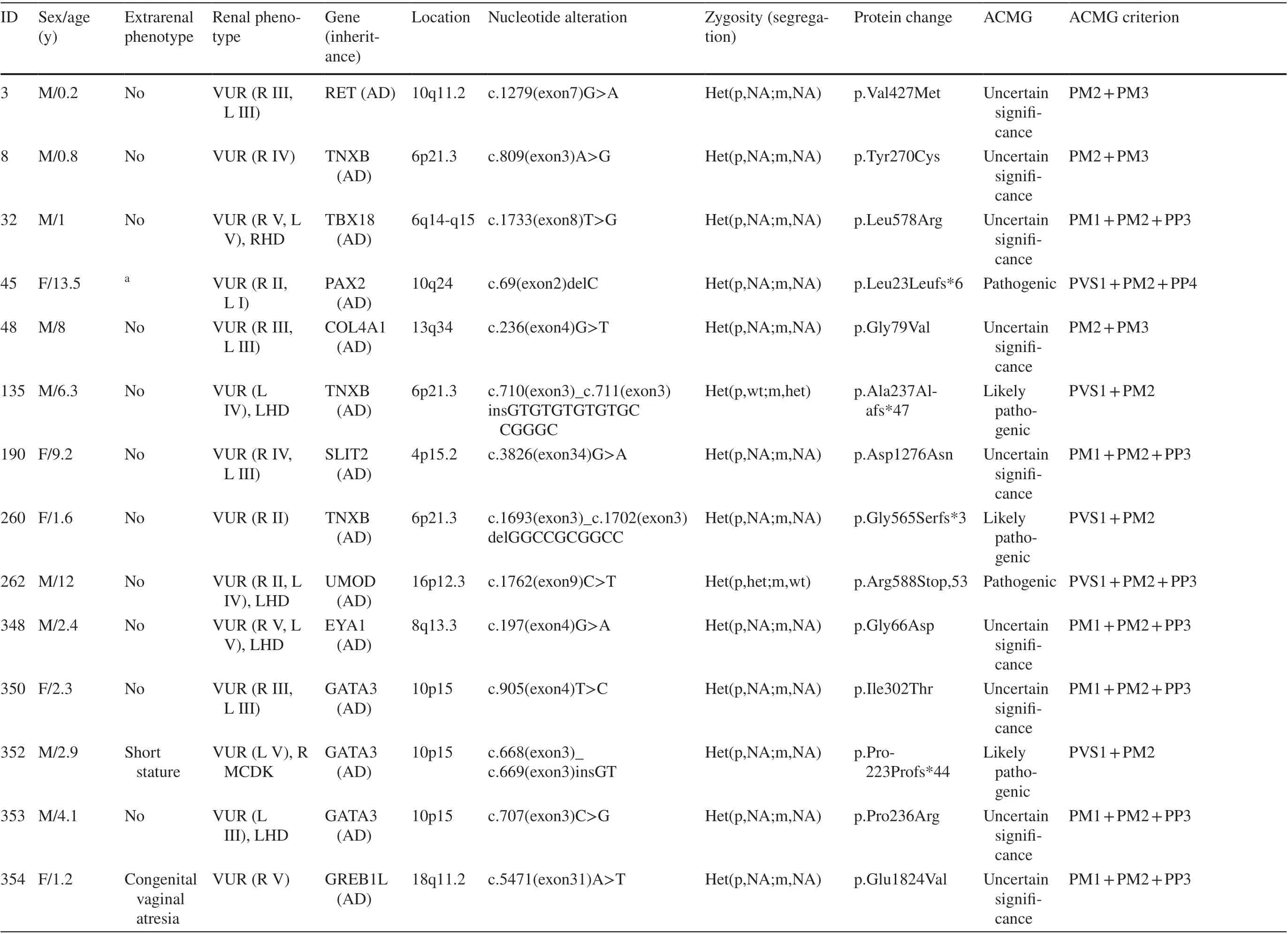
Table 1 Information on identified mutations in CAKUT genes or mutations in genes known to phenocopy CAKUT
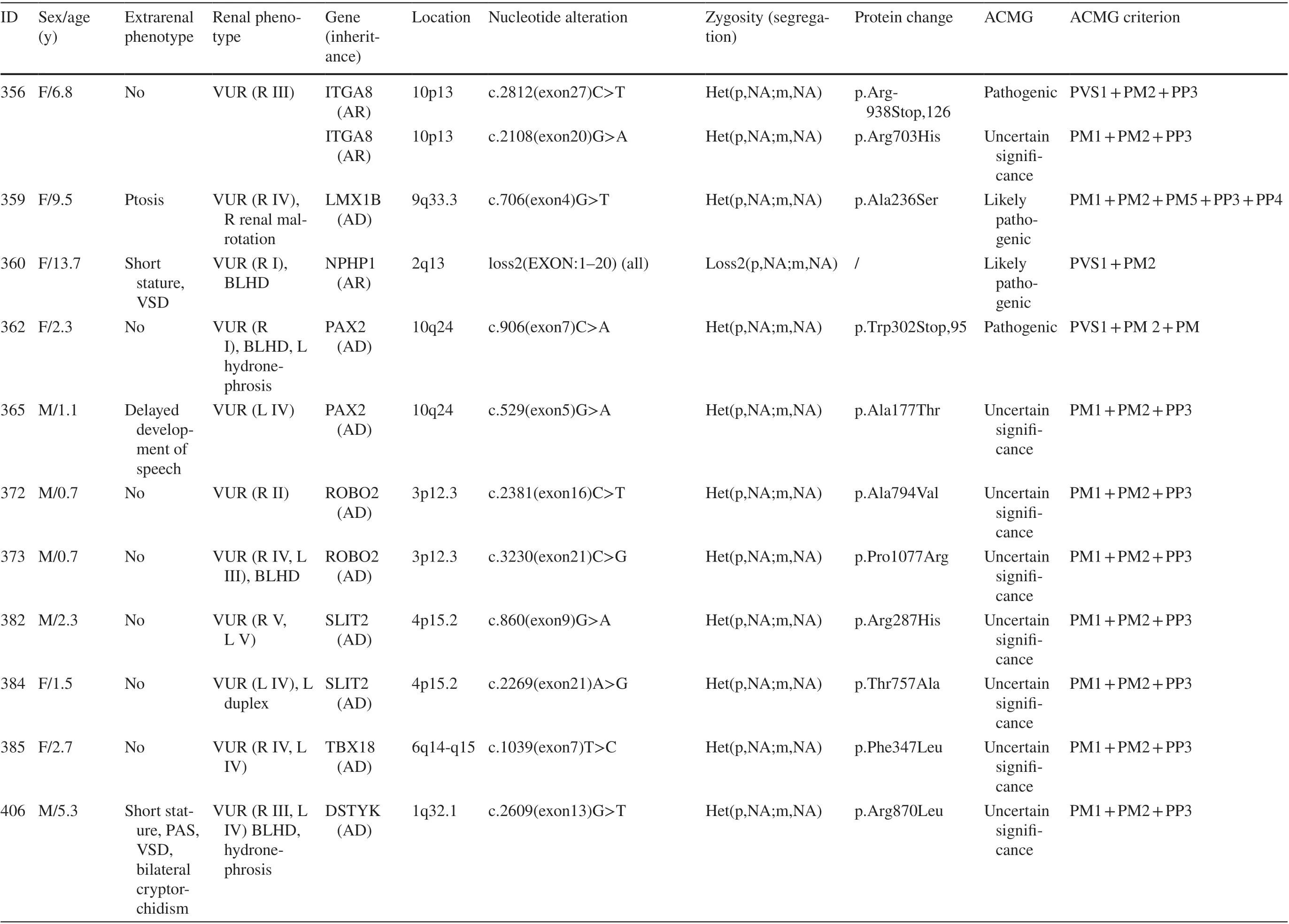
Table 1 (continued)

Table 1 (continued)

Table 2 Comparison between patients with and without gene variants

Fig. 2 a Log-rank test was used to compare the renal survival difference between the gene mutation and non-gene mutation group ( P = 0.010); b Log-rank test was used to compare the renal survival difference among the gene mutation groups ( P < 0.001)
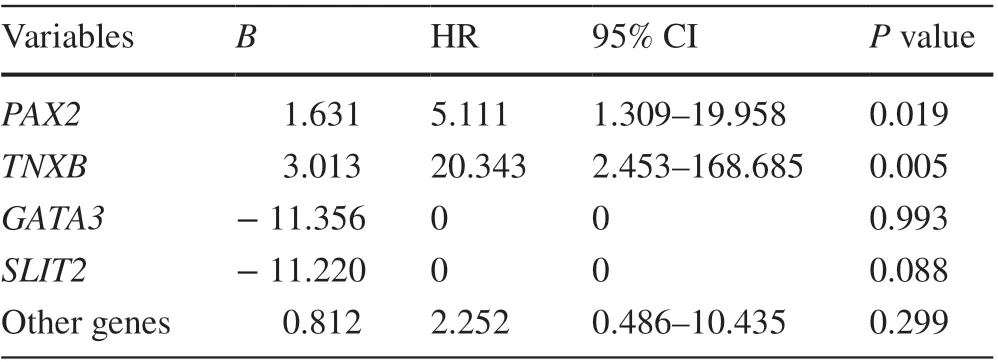
Table 3 Multivariable Cox regression analysis of candidate mutation in VUR patients
GATA3,HNF1B,TBX18,COL4A1,SALL1,SOX17,SIX5,TRAP1, andWnt4a[ 8- 10, 13, 15, 16, 20, 21] (Table 4).We detected the first 8 genes in our patients.PAX2, TNXB,GATA3andSLIT2were the main underlying monogenic causes of primary VUR in our study population (account for up to 46.4% of monogenic VUR). The most frequently mutated gene in this study wasPAX2, which accounted for 4 unrelated individuals with VUR and is similar to previous findings [ 9, 10, 14]. Mutations inPAX2cause the autosomaldominant disorder renal coloboma syndrome. Two patients withPAX2mutations in our study had ophthalmological abnormalities of the optic disc. The fact that we and previous studies [ 10, 14] did not identify mutations inSOX17,
BMP4,SIX1, orUPK3Asuggests that mutations in these genes are rare.
The genetic background of primary VUR has been investigated for many years. The identification of the genetic architecture of VUR is difficult because of the phenotypic heterogeneity and multifactorial genetic penetrance [ 9]. It is difficult to accurately determine whether the parent carrying the same mutation had primary VUR because many patients are asymptomatic, and it resolves spontaneously with age.
The enormous variability in detection rate (1-21%)[ 8- 10, 13, 15, 16, 20, 21] of genetic causes in different studies depends on methodological differences, e.g., the different methods of genetic testing, such as single-gene
screening, gene panel, next-generation sequencing (NGS),copy number variations (CNVs) analysis and chromosomal analysis [ 22, 23] (Table 4). Other factors are differences in family history and inclusion/exclusion criteria. CAKUT phenotypes are uncertain because voiding cystourethrograms (VCUGs) are not always performed. The same concerns exist for the diagnosis of extrarenal complications.Other reasons that may explain the different frequencies include differences in ethnicity and gender. Fetal exposure to teratogens and environmental toxicants may also be a factor [ 24].
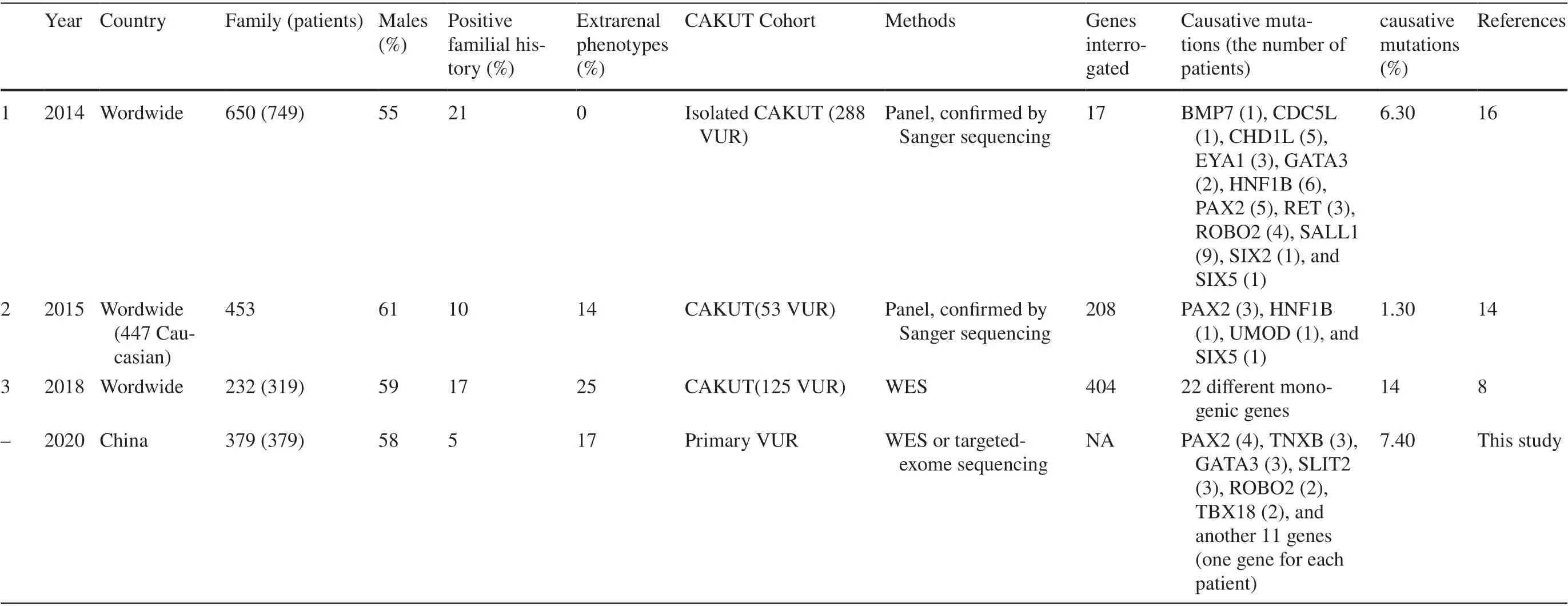
Table 4 Summary of molecular genetic studies (more than 50 participants) of VUR
Extrarenal manifestations may provide an important clue for uncovering the genetic cause of VUR [ 24]. We examined whether the detection rate of genetic causes differed between syndromic and non-syndromic VUR and recommend that genetic analysis be considered in syndromic cases. This study has shown thatPAX2mutation andTNXBmutation is significantly associated with earlier ESRD. Renal survival did not differ regardless of the presence ofGATA3mutation andSLIT2mutation.
Molecular diagnosis helps identify subtle clinical manifestations of other organs associated with genetic defects to initiate appropriate therapeutic strategies and affect prognosis of patients.
Further studies are necessary to understand the genotype/phenotype correlations and the genetic bases of long-term outcomes.
In conclusion, our study showed that a monogenic cause was discovered in 7.3% patients with primary VUR.PAX2,TNXB,GATA3andSLIT2were the main underlying monogenic causes and accounted for up to 46.4% of monogenic VUR. Extrarenal complications and renal function were significantly related to the findings of the genetic factors in children with primary VUR. Similar to other types of CAKUT,several genes may be responsible for isolated VUR. Our results confirm the previous conclusion that VUR is genetically heterogeneous. Although many genes did not appear to play a major role in isolated, non-syndromic VUR, these genes must be considered because the rate of gene mutations was similar to other types of CAKUT. There are several limitations associated with this study. First, we may have missed variations in noncoding regions of tested genes and genes involving other regions (CNVs and chromosomal abnormality) that were not covered. Second, triplicate samples were not available for all patients, and the assessment of segregation was not sufficient.
Supplementary InformationThe online version contains supplementary material available at https:// doi. org/ 10. 1007/ s12519- 021- 00428-x.
AcknowledgementsWe thank all participating patients and their families. Also, we thank our coordinators from Chigene (Beijing), Translational Medical Research Center Co. Ltd., WuXi NextCODE’s Shanghai, Beijing MyGenostics Co. Ltd., for sequencing technology support.
Author contributionsJLL, QS, MYW, GHZ, YFL are joint first authors. The roles of JLL, QS, MYW, GHZ and YFL are data curation, formal analysis, investigation, methodology, validation, visualization, writing-original draft, writing-review and editing. The roles of XWW, XST, YLB, YNG, JC, XYF, YHZ, GML, YBS, XJG, CHL,XYJ, SH, YLK, YLG, LPR, DL and SW are investigation and data curation. The role of BBW, DM, JR, QS and HX are writing-review and editing. The role of JLL and HX is funding acquisition. The roles of QS and HX are conceptualization, resources, supervision and project administration. All authors have seen and approved the final version of the manuscript.
FundingThis work was supported by the Grant NSFC-81800602 from National Natural Science Foundation of China (Dr. Jia-Lu Liu); the Grant 20184Y0176 from Shanghai Municipal Commission of Health and Family Planning Youth Research Program (Dr. Jia-Lu Liu); the Grant SHDC12016107 from Shanghai Shenkang Hospital Developmental Center (Dr. Hong Xu); the Grant NSFC-81670609 from National Natural Science Foundation of China (Dr. Hong Xu); the Grant 2018YFA0801102 from National Key Research and Development Project (Dr. Hong Xu).
Declarations
Ethical approvalThe study was approved by the Children’s Hospital of Fudan University’ Ethical Committee (no. 2017-56), and informed consent was obtained for all research individuals.
Conflict of interestAuthor Hong Xu is a member of the Editorial Board forWorld Journal of Pediatrics. The paper was handled by the other Editor and has undergone rigorous peer review process. Author Hong Xu was not involved in the journal’s review of, or decisions related to, this manuscript. All the other authors have no conflicts of interest to disclose.
 World Journal of Pediatrics2021年4期
World Journal of Pediatrics2021年4期
- World Journal of Pediatrics的其它文章
- Increased asprosin is associated with non-alcoholic fatty liver disease in children with obesity
- Mosaic trisomy 12 diagnosed in a female patient: clinical features,genetic analysis, and review of the literature
- Increasing prevalence and influencing factors of childhood asthma:a cross-sectional study in Shanghai, China
- Pediatric interfacility transport effects on mortality and length of stay
- Impact of probiotics supplement on the gut microbiota in neonates with antibiotic exposure:an open-label single-center randomized parallel controlled study
- Characteristics of immune and inflammatory responses among different age groups of pediatric patients with COVID-19 in China