
Impact of probiotics supplement on the gut microbiota in neonates with antibiotic exposure:an open-label single-center randomized parallel controlled study
2021-11-06 01:00:28HuiZhongXiangGengWangJingWangYanJieChenHuanLongQinRongYang
World Journal of Pediatrics 2021年4期
Hui Zhong 1 · Xiang-Geng Wang 1 · Jing Wang 1 · Yan-Jie Chen 1 · Huan-Long Qin 2 · Rong Yang 1
Abstract
Keywords Antibiotics · Gut microbiota · Neonatal infection · Probiotics
Introduction
Gut microbiota are established during the neonatal period and coevolve with the host to maintain long-term health [ 1].A healthy and stable community of gut microbiota serves various useful functions in several processes throughout the life of an individual, such as the development of the immune system, nutrient absorption, regulating the intestinal structures, the metabolism, and protecting against colonization by pathogens [ 2]. Microbial imbalance exerts adverse effects on the host, known as dysbiosis. Although the composition of adult gut microbiota is relatively stable, the microbial composition in the gut of a developing infant is highly dynamic.From birth onwards, the development of this microbial community is regulated by complex interactions between the host and the environment [ 3]. Several studies have demonstrated an association between disturbances to the gut microbiota during infancy and early childhood with health disorders, such as obesity, asthma, metabolic syndromes,chronic inflammatory diseases, antibiotic associated diarrhea, and opportunistic pathogen infection [ 4- 7].
Various factors, such as the mode of delivery, type of feeding, antibiotic use, and geographic factors, can affect the colonization and maturation of the gut microbiota in infants [ 8]. In particular, the perturbation of the developing infant gut microbiota by antibiotics has been linked to increased health problems in later life [ 9- 12], which is thought to occur, because altering the microbial community disturbs the immune and metabolic pathways [ 13]. Equally important is the reservoir of antibiotic resistance genes and the production of highly resistant bacterial strains [ 14]. In addition, antibiotic exposure can eliminate non-pathogenic commensal bacteria, allowing opportunistic pathogens,such asClostridium difficile, to bloom. Supplementation with probiotics has become increasingly popular as a means of alleviating the loss of gut microbial diversity [ 15]. The consumption of probiotics in the context of disease has received a large amount of public enthusiasm that greatly exceeds evidence of the efficacy obtained via research. As live microbes, probiotics show health promoting benefits for the host, mainly the generaLactobacillus,Lactococcus,orBifidobacterium. However, a lack of clear-cut guidelines on when to utilize probiotics and the most effective probiotic for different health conditions may be confusing for both physicians and patients [ 16]. The present study attempts to explore the impact of probiotics on the diversity of the intestinal microbiota of neonates when taken simultaneously with antibiotic treatment or throughout the recovery phase following antibiotic treatment. A cohort study was established to observe the potential protective benefits of probiotics on the gut microbiota of neonates that received antibiotic treatment.Evidence of these benefits was obtained with 16S rRNA sequencing.
Methods
Randomization of enrolled neonates and treatment strategies in the three groups
This study was performed at the Department of Pediatrics, Shanghai Tenth People’s Hospital, Tongji University School of Medicine between February 2017 and December 2018. Ninety enrolled neonates were diagnosed with bacterial infection by two independent pediatricians and treated with antibiotics with the informed consent of their guardians within 3 days of birth. A restricted block randomization sequence was created with a 1:1:1 allocation using a fixed block size of six. The block size was unknown to both investigators and participants. A data manager who was not associated with the clinical portion of this study prepared the randomization sequence using computer-generated random numbers. The inclusion criteria were as follows:(1) gestational weeks ≥ 37 weeks, but < 42 weeks; (2)birth weight ≥ 2500 and < 4000 g; and (3) white blood cell count ≥ 30 × 10 12 /L or C-reactive protein ≥ 10 mg/L.The exclusion criteria were: (1) mother received antibiotic treatment during delivery; (2) mother had fever before delivery; (3) mother had positive result for prenatal group BStreptococcusduring screening; (4) infant had a history of asphyxia (with 1, 5, and 10-minute Apgar scores ≤ 7) at birth; (5) infant had congenital malformation of the digestive tract or diseases of the liver, cholecyst, or pancreas;and (6) infant had congenital metabolic or hereditary disease. The enrolled neonates were then randomized into three groups: NI (no intervention, antibiotic therapy only), PCA(probiotics used concurrently with antibiotics), and PAA(probiotics used after antibiotics). The NI group received antibiotic treatment for 1 week, the PCA group received antibiotic treatment together with probiotics for 1 week,and the PAA group received treatment with antibiotics for 1 week followed by probiotics for 1 week. The antibiotic piperacillin-tazobactam (100 mg/kg/day) was administered twice per day. The probiotic supplement (BIFICO, Shanghai Sinepharm, China, half bag per treatment, three times per day) comprised > 1.0 × 10 7 CFU ofBifidobacterium longum,Lactobacillus acidophilus, andEnterococcus faecalis. Informed consent was obtained from the guardians of the enrolled neonates. This protocol was approved by the Ethics Committee of Shanghai Tenth People’s Hospital and adhered to the tenets of the Declaration of Helsinki.Informed consent to participate in the study was obtained from the legal guardian. This study was registered at Clinicaltrials.gov (NCT03388112).
Fecal sample collection of the neonates
Fecal samples were collected at four points in time; newborn(T0), 1 week (T1), 2 weeks (T2) and 42 days (T3) after birth,and were stored at - 80 °C in a refrigerator for further analysis. Clinical information to be used in the analysis, including the delivery mode, sex, and birth weight, were retrieved from the digital medical records system.
DNA extraction, sequencing, and bioinformatics analysis
The procedures used in this study were described in a previous study [ 17]. Briefly, DNAs were extracted from fecal samples using the E.Z.N.A.® Soil DNA kit (Omega Biotek, Norcross, GA, USA) according to the manufacturer’s instructions and were quantified using a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Fisher Scientific,Wilmington, MA, USA). The hypervariable V3-4 regions of the 16S rRNA gene in the gut microbiota were amplified by polymerase chain reaction (PCR) using specific primers and sequenced. PCR was performed under the following conditions: 95 °C for 3 minutes, followed by 27 cycles at 95 °C for 30 seconds and 55 °C for 30 seconds, with a final extension at 72 °C for 10 minutes. The amplified 16S rRNA amplicons were then purified using a DNA gel extraction kit (Axygen Biosciences, Union City, CA, USA) and sequenced using the Illumina MiSeq platform (Illumina, San Diego, CA, USA).The raw FASTQ files were demultiplexed, quality-filtered with Trimmomatic, and merged with FLASH. The operational taxonomic units (OTUs) were clustered at 3% divergence (97% similarity). Chimeric sequences were identified and removed using Uchime (version 4.2.40; http:// drive5.com/ usear ch/ manual/ uchime_ algo. html ). A taxonomic analysis of the representative sequences for each OTU was performed. The RDP Classifier algorithm ( http:// rdp. cme.msu. edu/) was used to analyze the taxonomy of each 16S rRNA gene sequence. Richness and diversity comparisons of the microbial community were performed after OTU identification. The taxa that were differently enriched in each group were identified using linear discriminant analysis coupled with effect size (LEfSe). Differences in the microbial structure were evaluated by principal coordinates analysis(PCoA). The number of permutations used to compare the microbial differences was set to 999. The Cytoscape platform (version 3.4.0; http:// www. cytos cape. org/) was used for co-abundance analysis. Function prediction and annotations were conducted using the Kyoto Encyclopedia of Genes and Genomes databases, the phylogenetic classification of proteins encoded in complete genomes, and the phylogenetic investigation of communities via reconstruction of the unobserved states.
Statistical analysis
Continuous variables were expressed as mean ± standard deviation. One-way analysis of variance was used to evaluate differences in the continuous variables. Chi-square tests were used to assess categorical variables. Mann-WhitneyUrank tests were used to compare the differences in the compositions of gut microbiota for two groups. Pearson and Spearman correlation tests were used to test for a relationship between bacterial genera and biochemical markers,where appropriate. All statistical analyses were performed with a PASW SPSS 22.0 (IBM, New York, NY, USA) and GraphPad Prism 7.00 (San Diego, CA, USA). Differences were considered to be significant whenP< 0.05. All comparisons were corrected using the Bonferroni method.
Results
The characteristics of enrolled neonates in the present study
Of the 90 neonates that were originally enrolled in this study,only 55 were included in the analysis based on our inclusion and exclusion criteria. Seventeen neonates and 68 fecal samples were included in the NI group with 25 neonates and 100 fecal samples from the PCA group and 13 neonates with 52 fecal samples from the PAA group. The baseline characteristics of the neonates are listed in Table 1. All enrolled neonates had Apgar scores > 7 at 1, 5, and 10 minutes with no need for resuscitation. None of the included neonates had positive blood cultures. Fifteen neonates were diagnosed with neonatal pneumonia, five neonates with urinary tract infection, and the remaining 35 neonates with non-specific infection. No neonates were diagnosed with neonatal purulent meningitis, and none showed gastrointestinal symptoms, such as vomiting, abdominal distension, diarrhea, or hematochezia, with negative fecal routine and fecal culture.None of the neonates were subjected to invasive or noninvasive ventilation, gastric or urethral catheterization, or deep venous catheterization.
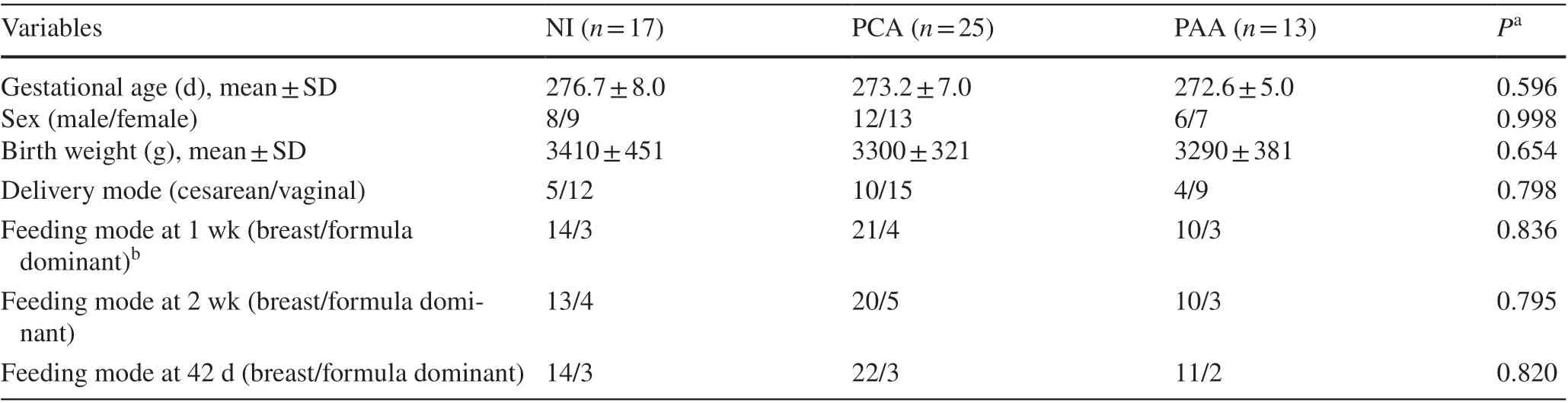
Table 1 Demographic characteristics of neonates in the study
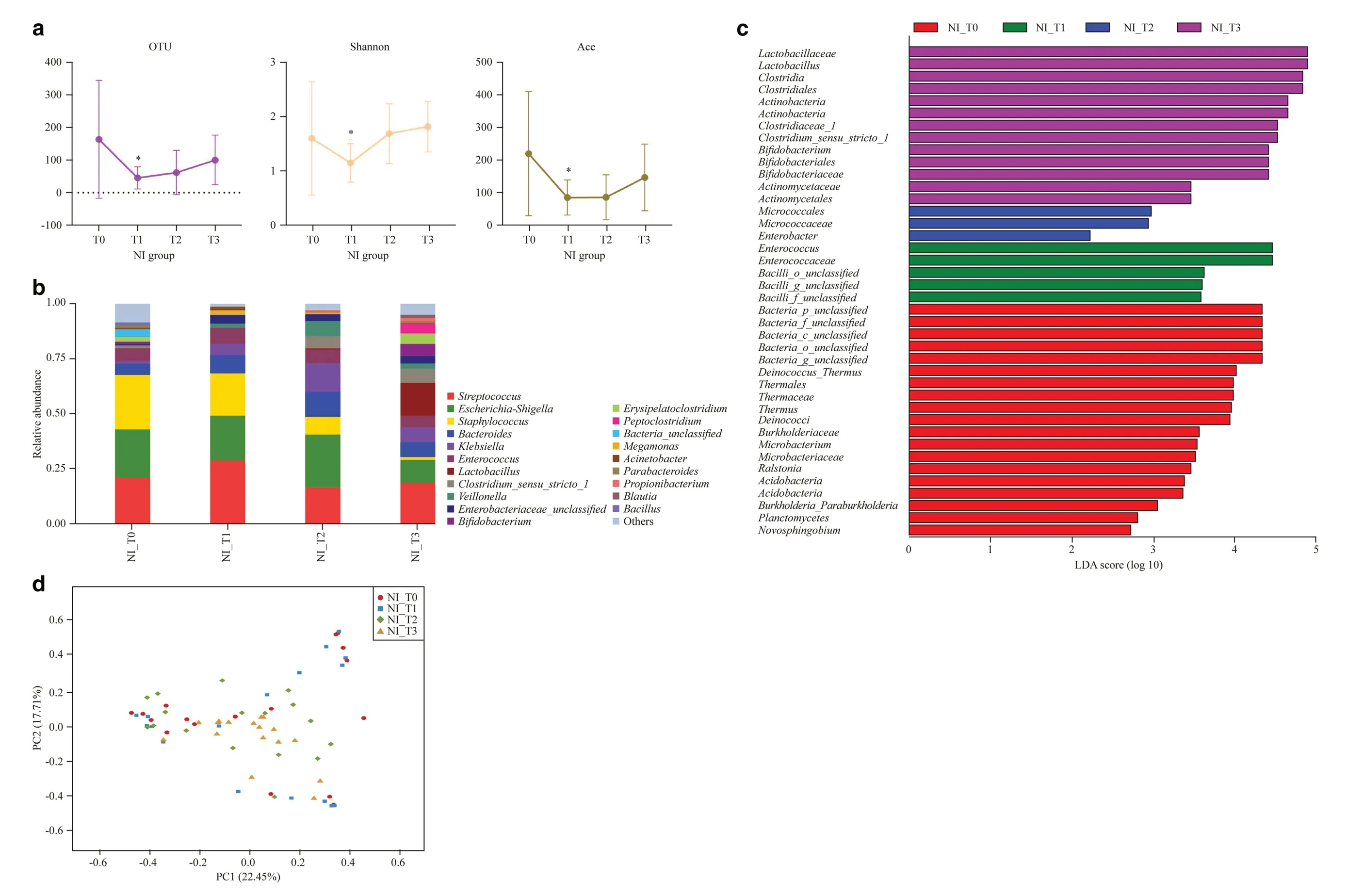
Fig. 1 Overview of gut microbiota at four time nodes in NI group. a Number of OTUs, and the Shannon and Ace indices at T0, T1, T2, T3, * P < 0.05, T1 vs. T0; b dominant genera in the NI group; c linear discriminant analysis effect size method showing differentially abundant genera; d unweighted principal co-ordinate analysis scores based on the relative abundance of OTUs (97% similarity level). Each symbol represents a sample. OTU operational taxonomic units, NI no intervention, T0 newborn, T1 1 week, T2 2 weeks, T3 42 days after birth, PC principal component score
Impact of antibiotic treatment on the gut microbiota of neonates in NI group
The 16S rRNA gene is universally present across all bacteria and is highly conserved, meaning that it is easily amplified using universal primers. Nine hypervariable regions can be used to distinguish between different organisms or species.The V3-V4 region was selected for the 16S rRNA amplicon analysis as it is commonly used in this type of process.The number of OTUs and Shannon and Ace indices were observed to decrease significantly at node T1 in the NI group as compared to node T0 (Fig. 1 a). Despite a slowly increasing trend in diversity after discontinuation of the antibiotic,the number of OTUs and the Ace index remained relatively low at node T3 than at T0, although the differences were not statistically significant (Fig. 1 a). These results suggest that antibiotic exposure significantly influences the initial development of the gut microbiota in neonates.
Hierarchical clustering analysis was used to define the microbial composition of the NI group at the four time nodes.At the phylum level,Actinobacteriadecreased significantly after treatment with antibiotics for 1 week (T0 vs. T1: 2.19%vs. 0.27%,P< 0.05) (Supplementary Fig. 1). At the genus level, the abundance of some bacteria includingBifidobacterium, Erysipelatoclostridium, Blautia, Lactobacillus,Clostridium_sensu_stricto_1, Peptoclostridium, and Propionibacteriumfluctuated significantly following treatment with the antibiotic; however, the abundance of these bacteria gradually increased with time in the later nodes (Fig. 1 b).Staphylococcus, Parabacteroides,andBacillusshowed decreasing trends in abundance over time (Fig. 1 b). Notably, colonization withBifidobacterium(T0 vs. T1: 1.18% vs.0.08%,P< 0.05) andLactobacillus(T0 vs. T1: 0.22% vs.0.01%,P< 0.05) decreased markedly after antibiotic treatment. Using the LEfSe method,Enterococcuswas identified as the most important bacteria after treatment with the antibiotic for 1 week.Lactobacillus and Clostridiawere dominant at 42 days, followed byActinobacteria, Clostridiaceae_1,Bifidobacterium,andActinomycetales(Fig. 1 c). Unweighted PCoA revealed differences in the composition of the bacterial community of the NI group at the four time nodes using the first two principal component scores PC1 and PC2 (22.45 and 17.71% of the explained variance, respectively) (Fig. 1 d).These results indicate that early life exposure to antibiotics has a detrimental impact on the colonization of some key bacteria, such asBifidobacterium,in the infant gut.
Impact of concurrent or heterochronous probiotics supplement on the gut microbiota of antibiotic-treated neonates
To assess the effect of administering probiotics concurrently with antibiotic treatment on the colonization of gut microbiota in early life, we compared the total bacteria of newborns (T0), 1 week (T1) and neonates 42 days after birth(T3) in the PCA group with those in the NI group. No statistically significant differences were observed between the number of OTUs or Shannon and Ace indices in the two groups (Supplementary Fig. 2a, b). Further comparison of the taxonomic alternations of the gut microbiota at the phylum and genus levels demonstrated a significant increase in the relative abundance ofActinobacteria(NI_T1 vs.PCA_T1: 0.27% vs. 1.01%,P< 0.05; NI_T3 vs. PCA_T3:9.08% vs. 11.1%,P< 0.05) andProteobacteria(NI_T1 vs.PCA_T1: 31.1% vs. 40.7%,P< 0.05; NI_T3 vs. PCA_T3:22.2% vs. 34%,P< 0.05) after probiotic supplementation at T1 and T3 (Supplementary Fig. 2c). Some potentially beneficial bacteria, such asBifidobacterium(NI_T1 vs. PCA_T1:0.08% vs. 0.18%,P< 0.05; NI_T3 vs. PCA_T3: 5.57% vs.7.54%,P< 0.05), were more abundant after using probiotics concurrently with antibiotic treatment (Fig. 2 a, b).
No significant difference between the number of OTUs or Shannon and Ace indices in the NI group and the PAA group was observed at any node when probiotic supplements were administered after antibiotic treatment (Supplementary Fig. 2a, d). At the phylum level,Actinobacteriaincreased significantly at T2 and T3 (NI_T2 vs. PAA_T2: 1.89% vs.7.32%,P< 0.05; NI_T3 vs. PAA_T3: 9.08% vs. 13.71%,P< 0.05) following supplementation with probiotics (Supplementary Fig. 2e). At the genus level,BifidobacteriumandLactobacillusshowed a transient and insignificant increasing trend at T2 when probiotic supplements were administered for 1 week after receiving antibiotic treatment, but no significant difference was observed at T3 (Fig. 2 c, d). This result indicates that delaying the administration of probiotic supplements did not benefit the “devastation” to the gut microbiota that resulted from antibiotic treatment.
Microbiota comparison of antibiotic-treated neonates receiving concurrent or heterochronous probiotic intervention
Fig. 2 Comparison of concurrent or heterochronous probiotics supplement on the gut microbiota of antibiotic-treated neonates. Dominant genera ( a, b) at T1 and T3 in the NI and PCA groups. The dominant genera ( c, d) at T2 and T3 in the NI and PAA groups. NI no intervention, PCA probiotics used concurrently with antibiotics, PAA probiotics used after antibiotics, T1 1 week, T2 2 weeks, T3 42 days after birth
In this part of the study, we further observed the impact of supplementation with probiotics at different stages of the antibiotic treatment on the microbial community structure of infants 42 days after birth. The comparison of the microbiota in the PCA and PAA groups was conducted at node T3. No significant difference was observed in the number of OTUs, and the Shannon and Ace indices for the two groups at T3 were also similar (Supplementary Fig. 3a).At the phylum level,Firmicuteswere the predominant gut microbiota, with a similar abundance observed in both the PCA and the PAA group (Supplementary Fig. 3b). At the genus level,Escherichia-Shigella, Streptococcus, Bifidobacterium, Bacteroides, Klebsiella, Veillonella, Staphylococcus,Peptoclostridium, Ruminiclostridium_5, Eisenbergiella,Erysipelatoclostridium, Enterobacteriaceae_unclassified,Subdoligranulum,andHaemophiluswere more abundant in the PCA group, whereasLactobacillus, Enterococcus,Clostridium_sensu_stricto_1, Propionibacterium, Rhodococcus,andActinomyceswere more abundant in the PAA group (Fig. 3 a). A bubble chart was used to show the relative abundance of genera in the three groups at 42 days (Fig. 3 b).Markedly,Bifidobacterium(PCA_T3 vs. PAA_T3: 7.54%vs. 3.40%,P< 0.05) was more abundant in the PCA group(Fig. 3 b). By comparing the PCA group with the PAA group at T3,γ-Proteobacteriawas identified as the key type of bacteria in the PCA group (Supplementary Fig. 3c). PCoA revealed that the first two principal component scores of PC1 and PC2 were 19.87 and 11.55%, respectively (Supplementary Fig. 3d).
Discussion

Fig. 3 Comparison of gut microbiota between PCA and PAA group. a Dominant genera in the PAA and PCA groups at T3; b prevalent bacterial genera identified in the three groups with relative abundance denoted by circle size and colors representing different phyla. NI no intervention,PCA probiotics used concurrently with antibiotics, PAA probiotics used after antibiotics, T1 1 week, T2 2 weeks, T3 42 days after birth
Antibiotics are very effective and are currently recommended for treating neonates with suspected clinical sepsis in accordance with the guidelines. Consequently, a considerable proportion of newborns are treated with antibiotics within the first few days of birth. One of the adverse effects of antibiotic treatment is the alteration of the composition of gut microbiota in newborns during the first weeks of life.The long-term clinical or microbiological impacts of this exposure remain unclear. One previous study demonstrated that use of antibiotics may disturb the establishment of the gut microbiome, especially in premature infants, and the composition of the microbiota is affected differently under the use of different antibiotics [ 18]. Another recent study reported that neonatal antibiotic treatment is associated with long-term disruption of the gut microbiota and led to the reduced growth of a boy over the first 6 years after birth [ 19].In the present study we found that exposure to a single antibiotic (piperacillin-tazobactam) can also decrease the richness of the gut microbiota in full-term infants and can disturb the reproduction ofBifidobacteriumandLactobacillus.Our results are consistent with previous observations [ 20,21], which have indicated a decreased abundance ofBifidobacteriumin infants that have been exposed to antibiotics.Epidemiological and experimental studies have suggested that neonatal antibiotic exposure might affect growth and lead to obesity and metabolic disease [ 22, 23]. Moreover,disturbance of the symbiotic flora with antibiotics may have adverse effects in the establishment of the immune system and lead to allergic diseases. The gut microbiota plays a vital role in T-cell differentiation in infants [ 24], and the reduced exposure to bacteria in early life inhibits the transformation from the T helper cell Th2 to Th1, resulting in an increase in the incidence of allergic diseases. The lack of certain bacteria may, therefore, result in allergies and asthma [ 25]. The use of antibiotics in premature infants has also been significantly associated with an increase in the incidence of allergic dermatitis, recurrent wheezing, and asthma at age six [ 26].The potential causal link between antibiotic exposure and these diseases may be mediated by antibiotic-induced perturbations of the developing gut microbiota.
Several studies have demonstrated a clear benefit in administering probiotics for routine prophylaxis to decrease the risk of necrotizing enterocolitis, sepsis, and antibiotic-associated intestinal dysbiosis in neonates [ 27,28]. Probiotics may restore the gut microbiota and introduce beneficial effects to the gut microbial community, resulting in the amelioration or prevention of gut inflammation and other intestinal diseases [ 29, 30]. However, a better understanding of how the gut microbiota can be restored effectively in patients would have significant clinical implications. One animal study reported that probiotic bacteria do not appear to colonize and become part of the gut microbiota, but probiotic supplementation did appear to significantly remodel the microbiome ofindividual mice that were recovering from antibiotic therapy [ 15]. However, the optimal timing, duration, and dosage of probiotic interventions have not been determined when used to treat several diseases in humans. To our knowledge, the present study was the first to focus on the timing of clinical intervention using probiotics following antibiotic treatment in the neonatal period. We investigated the impact of probiotics under two different conditions; concurrent with the antibiotic or sequential administration. We found that concurrent treatment with both antibiotics and probiotics had a highly significant impact,leading to an increase in the abundance ofBifidobacterium.There appears to be little benefit in treating gut microbiota that have been “devastated” by antibiotic treatment when supplementation with probiotics is delayed. The richness of this genus that results from probiotic supplementation could have an important influence on the ability of these bacteria to repopulate as part of the host gut microbiota, because supplementary probiotics can have far consequences in longtime health after antibiotic therapy. However, the mechanism is still largely unclear. Further research is required in terms of using probiotics for targeted microbial manipulation to determine the mechanisms by which a healthy gut microbiota can be promoted. In addition, it should be noted that concerns have been raised about the safety of probiotic supplementation in neonates, with problems, such as probiotic sepsis [ 31], the transmission of antibiotic resistance [ 32],the possibility of exaggerated pro-inflammatory reactions[ 33] and the difficulties of obtaining high-quality, safe and effective products [ 34, 35]. Although none of the neonates enrolled in our study developed systemic infections due to the use of probiotics, we must be aware that probiotic supplementation can cause sepsis in high-risk neonates on rare occasions, as documented in previous reports [ 36].
Despite our findings, there are still some limitations in our study. First, the sample is not of sufficient size to comprehensively clarify the effects of antibiotic exposure and supplementary probiotics on gut microbiota. Second,this study used 16S rRNA sequencing technology to detect gut microbiota, which could be further improved using technologies that are more accurate. Third, our follow-up did not cover a sufficient period for the long-term clinical or microbiological long-term effects of antibiotic exposure and supplementary probiotics to be comprehensively observed. These limitations are expected to be overcome using samples that are more clinical and well-designed,longer follow-up schemes.
In summary, our study showed that administering antibiotics to neonates leads to a decrease in the microbial richness and diversity of gut microbiota, with the attenuation of some bacteria, particularlyBifidobacteriumandLactobacillus.Although probiotics do not appear to alleviate this loss of diversity in the gut microbiota, they do help to reshape the gut microbiota as it repopulates. Compared to the delayed use of probiotics after treatment with antibiotics, the concurrent use of probiotics with antibiotics exerts more benefits to the gut microbiota, such as promoting the abundance ofBifidobacterium. These observations provide a rationale for caution when using antibiotics in clinical settings, and highlight the power of administering probiotic concurrently with antibiotics.
Supplementary InformationThe online version contains supplementary material available at https:// doi. org/ 10. 1007/ s12519- 021- 00443-y.
AcknowledgementsWe thank Majorbio Biological Technology Co.,Ltd. for providing technical assistance in this study. We also thank all the parents of the neonates participated in this study.
Author contributionsZH performed the collection, analysis, interpretation of data and wrote the first version of manuscript. WXG, WJ and CYJ helped collect the subjects’ information and samples. YR and QHL took responsibility for the integrity of the work as a whole from study inception to the published article. All authors read and approved the final manuscript.
FundingThis work was supported by grants from the National Natural Science Foundation of China (Nos. 81230057, 81200264, 81372615,and 81472262), the Emerging Cutting-Edge Technology Joint Research Projects of Shanghai (No. SHDC12012106), and the Tongji University Subject Pilot Program (No. 162385).
Compliance with ethical standards
Ethical approvalAll procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.This study was approved by the Ethics Committee of Shanghai Tenth People’s Hospital (approval no. SHSY-IEC-4.0/17-42/01). Informed consent to participate in the study have been obtained from their legal guardian.
Conflict of interestNo financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article. The authors have no conflict of interest to declare.
Data availabilityThe 16S sequence information in this study has been submitted to the NCBI Sequence Read Archive (SRA), with accession number SRP115062.
 World Journal of Pediatrics2021年4期
World Journal of Pediatrics2021年4期
- World Journal of Pediatrics的其它文章
- Increased asprosin is associated with non-alcoholic fatty liver disease in children with obesity
- Mosaic trisomy 12 diagnosed in a female patient: clinical features,genetic analysis, and review of the literature
- Increasing prevalence and influencing factors of childhood asthma:a cross-sectional study in Shanghai, China
- Responsible genes in children with primary vesicoureteral reflux:findings from the Chinese Children Genetic Kidney Disease Database
- Pediatric interfacility transport effects on mortality and length of stay
- Characteristics of immune and inflammatory responses among different age groups of pediatric patients with COVID-19 in China