
噻吩功能化聚苯乙烯的合成
2021-11-03 08:18:38李旭,牛慧,李杨
石油化工 2021年10期
李 旭,牛 慧,李 杨
(大连理工大学 化工学院 高分子材料系,辽宁 大连 116024)
聚苯乙烯(PS)因具有良好的电气性能、化学稳定性、机械性能和加工性能等优点,在包装、电子、建材、汽车内饰、家用电器和日用品等行业得到广泛应用[1-2]。随着现代科技的快速发展,高端应用领域对PS材料提出了更高要求,但是传统的PS高分子中缺乏功能化基团,影响了功能的提升和应用领域的拓展。对PS的改性通常包括共聚改性和共混改性,即将苯乙烯(St)与功能单体共聚或向PS中加入功能填料改善聚合物功能[3-5]。包括PS在内的大多数高分子材料均为绝缘体,在特定的应用环境下,赋予聚合物一定程度的导电性可以拓宽材料用途。提升聚合物导电性主要通过两种途径:1)在分子链结构中引入共轭结构以促进电子传输,但该方法往往使材料的加工性能变差;2)加入导电填料来构建电子通路,使材料在实际应用中兼具易加工性和导电性[6-10]。主链具有共轭结构的导电聚合物聚噻吩(PT)虽然具有优异的电学性能,但难加工的问题也极大地限制了它的应用范围[11-12],研究者目前主要通过在噻吩上引入烷基取代基等方法增加可加工性[13-19]。
本工作合成了一种噻吩功能化苯乙烯(Ts)单体,将其与St单体通过自由基聚合制备了噻吩功能化PS(PST),然后通过溴化反应及Suzuki偶联反应进一步提高聚合物中的噻吩基团数量,同时通过掺杂多壁碳纳米管(MWCNTs)的方法提高材料的导电性。采用1H NMR,GPC,DSC等方法考察了聚合物的组成、分子量及其分布、热性能和导电性能等。
1 实验部分
1.1 主要原料和试剂
2-噻吩甲醇、四丁基氢氧化铵、4-氯甲基苯乙烯、2,2'-偶氮双(异丁腈)(AIBN)、St、N-溴代丁二酰亚胺(NBS)、2-噻吩硼酸、四丁基溴化铵、四(三苯基膦)钯:纯度98%(w),萨恩化学技术(上海)有限公司;NaOH、无水碳酸钠:纯度98%(w),天津天大化学;甲苯、乙醚:分析纯,天津市大茂试剂厂;四氢呋喃(THF)、无水甲醇:分析纯,天津市富宇精细化工有限公司;MWCNTs:先丰纳米材料科技有限公司。
1.2 测试与表征
1H NMR表征采用Bruker公司AV 400 MHz型核磁共振波谱仪,以二甲基亚砜为溶剂、四甲基硅烷为内标、室温。MS表征采用Micromass UK Limited公司Q-TOF Micro型质谱仪。聚合物的分子量及其分布采用Waters公司2414型凝胶渗透色谱仪测定,溶剂THF、流量1.0 mL/min、测定温度30 ℃,并用标准PS试样进行校正。
采用TA公司Q2000型差示扫描量热仪进行DSC测试,N2环境,以10 ℃/min的速率从室温逐渐升温至200 ℃,在200 ℃下恒温5 min消除热历史,再以10 ℃/min 的速率降至0 ℃,最后以10℃/min的速率升至200 ℃,测试聚合物的玻璃化转变温度(Tg)。
采用广州四探针科技公司RTS-9型双电测四探针测试仪进行导电率测试,将试样在Tg以上20℃左右压成厚度为1 mm、直径1 cm左右的圆片,压片机压力为10 MPa,保压5 min,常温下测试。
1.3 单体Ts的合成
参照文献[20-21]报道的方法合成单体Ts,合成路线见式(1)。N2保护和室温下,在单支口瓶中加入212 mmol 2-噻吩甲醇和50 mL 33%(w)的NaOH水溶液搅拌3 h,然后向体系中加入溶于甲苯的106 mmol 4-氯甲基苯乙烯,最后加入21.2 mmol四丁基氢氧化铵,常温磁力搅拌下反应48 h后收集产物。后处理方法为:先向反应液中加入50 mL乙醚溶剂萃取有机产物,然后水洗有机相至下层液无色,加入无水MgSO4过夜干燥,过滤后使用旋转蒸发器将溶剂旋干得到Ts,产率为91%。1H NMR(400 MHz,C2D2Cl4)表征结果(化学位移δ)为:7.49(d,J=7.9 Hz,1H),7.45~7.30(m,1H),7.08(ddd,J=5.5,3.3,0.5 Hz,1H),6.90~ 6.70(m,1H),5.85(d,J=17.6 Hz,1H),5.35(d,J=10.9 Hz,1H),4.77(s,1H),4.61(s,1H);EI-MS:m/z=230。

1.4 聚合反应
采用自由基聚合法制备PST。N2保护下,在单支口瓶中加入17.4 mmol Ts、17.4 mmol St及引发剂0.05 g AIBN,再加入10 mL甲苯溶剂,60 ℃油浴加热反应24 h后收集产物;将产物滴入甲醇溶液中搅拌沉胶,洗去未反应的单体和引发剂,过滤之后将产物放入真空烘箱中50 ℃下过夜干燥,所得聚合物记为PST3(其余产品见表1),称重计算产率,密封保存。
PST的合成路线见式(2)。通过调整共聚单体配比,可合成一系列噻吩含量不同的PST(见表1)。还可对PST中的噻吩基团进行扩链:首先对噻吩基进行溴化反应,得到溴化聚合物PSTBr;再通过PST-Br与2-噻吩硼酸之间的Suzuki偶联反应,在共聚物中继续接入噻吩基,得到接枝产物PST-g-T,从而进一步提高PS的噻吩基团含量。本工作制备的所有聚合物均可在常温下溶于THF。
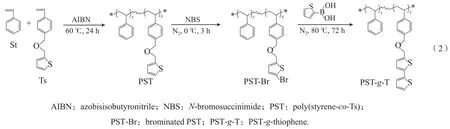
1.5 聚合物的溴化反应
聚合物的溴化反应参照文献[22]报道的方法。在氮气保护下,单支口瓶中加入3.8 mmol PST,然后加入3.8 mmol NBS,加入10 mL THF溶剂,在冰水浴下避光反应3 h,收集产物;将产物滴入甲醇溶液中搅拌沉胶,洗去未反应的NBS,过滤之后放入真空烘箱中50 ℃下过夜干燥,所得产物记为PST-Br,称重计算产率,密封保存。
PT的溴化产物记为PT-Br。
1.6 聚合物的接枝反应
采用Suzuki偶联反应,参照文献[23]报道的方法。在氮气保护下,单支口瓶中加入1.5 mmol PST-Br,再加入1.6 mmol 2-噻吩硼酸、0.03 g四(三苯基膦)钯、0.15 g四丁基溴化铵、3 mL 2 mol/L NaCO3水溶液,加入10 mL甲苯溶剂,在80 ℃油浴中加热72 h后收集产物;将所得产物过滤除去不溶物,溶液滴入甲醇溶液中搅拌沉胶,洗去未反应的物质,然后将产物过滤放入真空烘箱中50 ℃下过夜干燥,得到的接枝产物记为PST-g-T,称重计算产率,密封保存。
PT的噻吩接枝产物记为PT-g-T。
1.7 PST/MWCNTs复合材料制备
将PST溶于THF中,按一定质量比加入MWCNTs,充分搅拌混合后旋干溶剂,将混合后的产物放入真空烘箱中50 ℃下干燥过夜,得到PST/MWCNTs;干燥后将PST/MWCNTs在Tg以上20 ℃左右压成厚度为1 mm、直径1 cm左右的圆片,压片机压力10 MPa,保压5 min,待冷却后保存。
2 结果与讨论
2.1 PST的性能
将Ts与St按不同配比进行自由基共聚制备PST,反应参数及PST的组成与性能见表1。
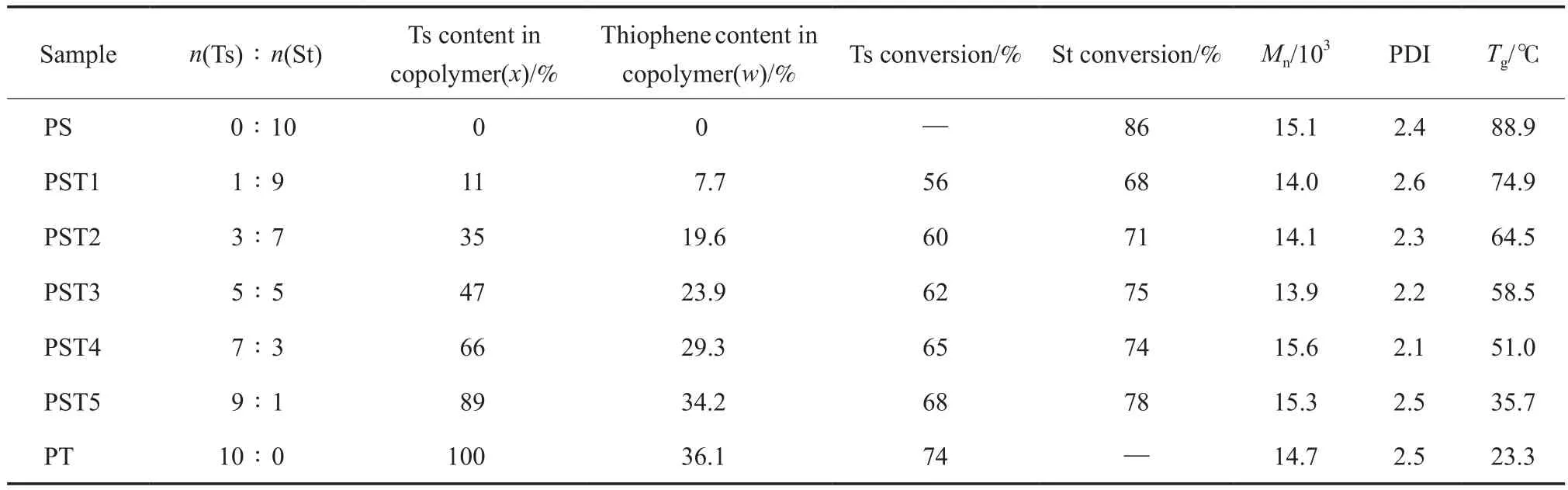
表1 PST的组成与性能Table 1 Composition and characterizations of PST
聚合物的1H NMR谱图见图1。从图1可看出,PST主链—CH—,—CH2—中H原子的δ=1.0~2.0,苯环中H原子的δ=6.2~7.3。单体Ts的特征峰有两处:1)与O原子相连的两个亚甲基,H原子的δ=4.6,4.8,聚合后偏移至4.3和4.5处;2)与S原子相连的亚乙烯基,H原子的δ出现在7.4处。上述两处特征峰在PT和PST中均清晰可见,而在PS的谱图中未出现。这表明Ts与St成功共聚。PST的组成可通过对δ=4.0~5.0处的Ts特征峰面积、结合δ=6.2~7.3处St与Ts共同特征峰(苯环)的面积进行计算,结果见表1。从表1可知,共聚物组成与单体配比相近,这说明可通过改变反应物配比对聚合物组成进行调节。St的转化率高,在68%~78%左右;Ts的转化率略低于St,但也保持在56%~68%较高水平。
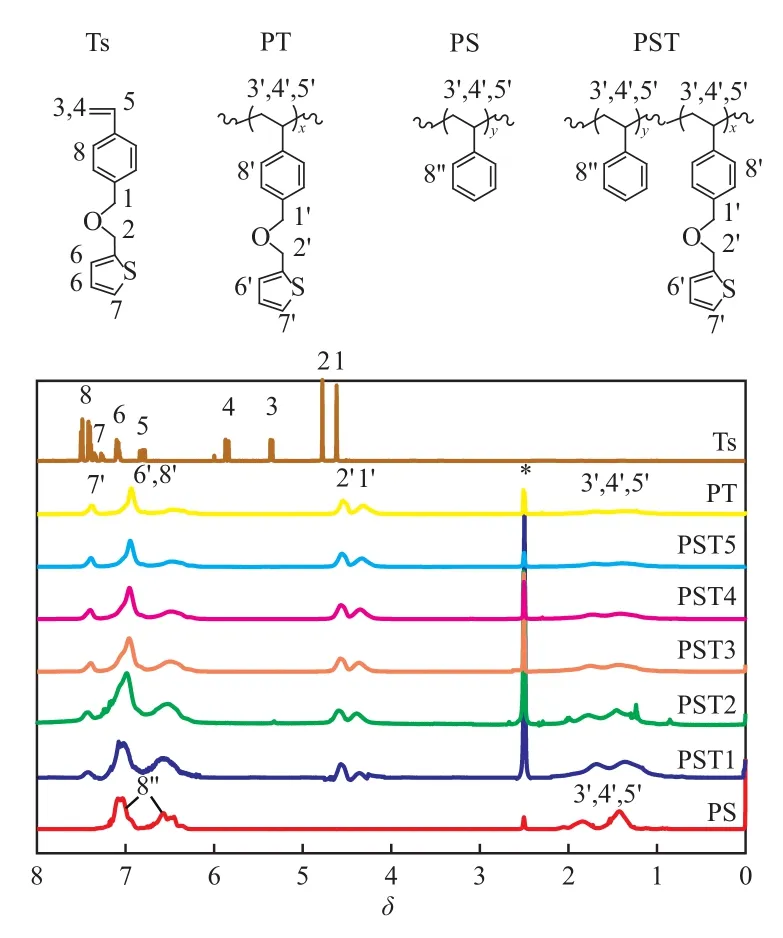
图1 PST的1H NMR谱图Fig.1 1H NMR spectra of the PST.
图2为聚合物的GPC曲线。从图2可看出,所有聚合物均呈单峰分布,说明聚合物分子量随组成变化不大。进一步对聚合物进行DSC测试,结果见图3。从图3可看出,不同的聚合物均出现单一的Tg,这表明PST为无规共聚物;并且Tg随PST中Ts含量的增加而逐渐下降,这可能与Ts单体中含有醚键有关,C—O—C的柔性赋予PST侧基柔性,且Ts含量越高,PST的Tg越低,均聚物PT的Tg已降至室温(23.3 ℃)附近。
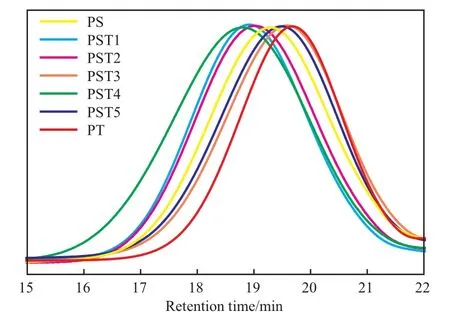
图2 聚合物的GPC曲线Fig.2 GPC curves of the polymers.

图3 聚合物的DSC曲线Fig.3 DSC curves of the polymers.
2.2 噻吩接枝反应
溴化和噻吩接枝聚合物的1H NMR谱图见图4。从图4可看出,δ=7.38处的特征峰几乎完全消失,表明该处的H原子已被Br取代。通过对该处残存的特征峰面积积分计算未反应的H原子含量,可得到溴化反应效率,结果见表2。从表2可知,溴化反应较完全,所有试样的溴化效率均在96%以上。溴化聚合物与2-硼酸噻吩通过Suzuki偶联反应,完成噻吩基在共聚物侧基的接枝。反应完成后,PST3-g-T和PT-g-T位于δ=7.4处的特征峰再次出现,同时位于δ=7.0左右代表噻吩环上H原子的特征峰明显增强,表明接枝成功。通过对该处特征峰面积积分,可以计算接枝反应效率(见表2)。从表2可知,所有试样的噻吩接枝效率均在90%以上,即聚合物中的噻吩含量进一步提高。

表2 溴化和接枝反应后聚合物组成和性能Table 2 Polymer composition and characterizations after brominating and grafting reaction
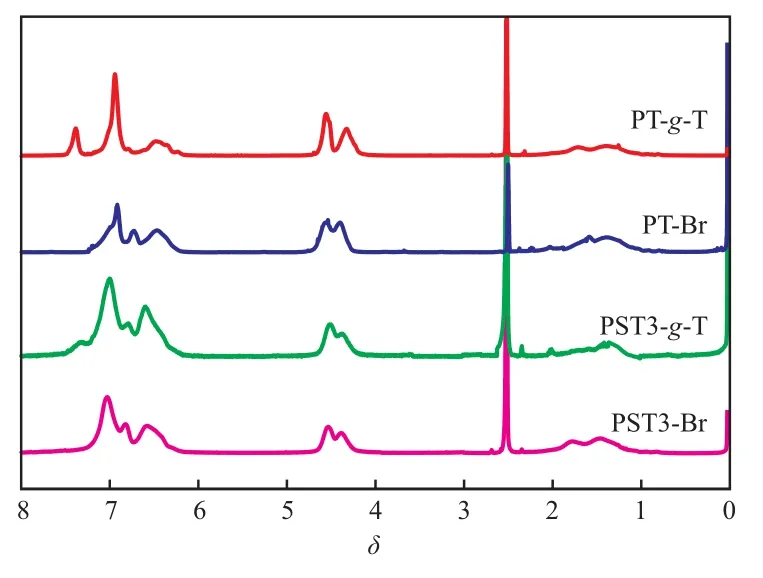
图4 溴化和噻吩接枝聚合物的1H NMR谱图Fig.4 1H NMR spectra of brominated polymers and thiophene grafted polymers.
溴化后和噻吩接枝后聚合物的GPC曲线见图5。从图5可看出,聚合物溴化后的GPC保留时间明显前移,表明溴化反应使聚合物分子量增加;同时分子量分布变宽,这可能是溴化效率不足100%导致。接枝后的聚合物GPC曲线保留时间进一步前移,证明Br原子被分子量更高的噻吩基取代。均聚物PT接枝后的GPC曲线前移程度大于PST3,这是由于PT的每个侧基都可进行接枝反应,而PST3仅有47%的侧基为可接枝Ts单元,其余为无法接枝的St单元。所有试样的GPC曲线均呈单峰,这说明溴化反应和噻吩接枝反应均可高效完成。
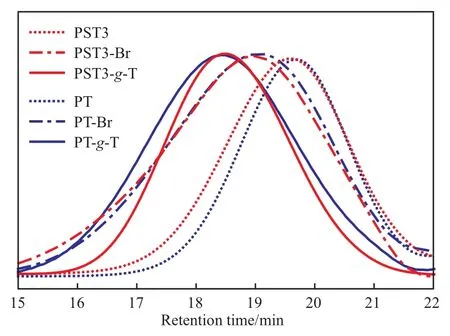
图5 溴化后和接枝后聚合物的GPC曲线Fig.5 GPC curves of the polymers after brominating and grafting reaction.
接枝聚合物的DSC曲线见图6。从图6可看出,接枝聚合物的Tg变化表现出与共聚物相反的趋势。噻吩含量较低的PST1接枝后所得产物PST1-g-T的Tg变化不大;而PST2~PST5接枝噻吩基团后,Tg显著提高,且随噻吩含量增加,Tg升高趋势增大。PT接枝噻吩得到的PT-g-T的Tg由接枝前的23.3℃增高至接枝后的110.9 ℃。可见,单体中两个相连的噻吩基即可使聚合物呈明显的刚性。通过逐步接枝噻吩基的方法,能够对PST的组成、热性能和加工性能进行逐步调控,使PST保持可溶解、可加工的优点。
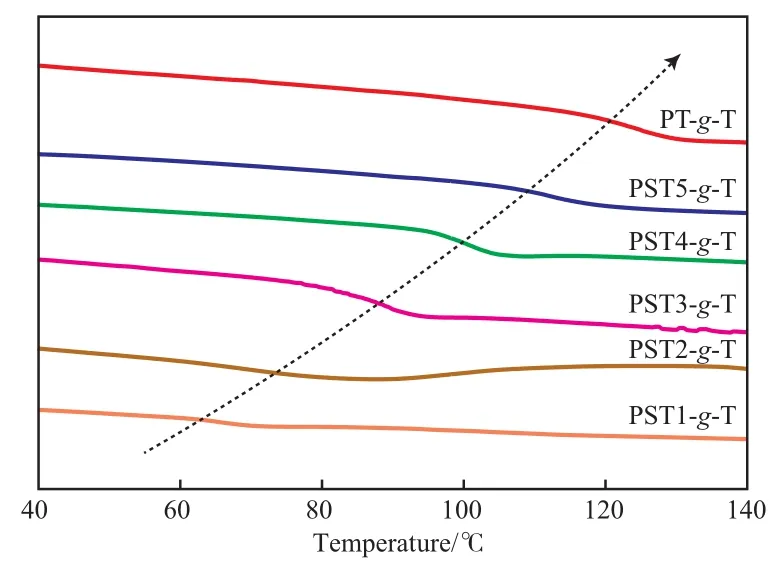
图6 接枝聚合物的DSC曲线Fig.6 DSC curves of the grafted polymers.
2.3 复合材料电导率
聚合物/MWCNTs薄膜的电导率见图7。从图7可看出,未掺杂MWCNTs的聚合物的电导率低于仪器测试范围;掺杂5%(w)MWCNTs的聚合物薄膜的电导率为0.1~1 S/m,随着MWCNTs掺杂量的增加,薄膜电导率迅速上升,在MWCNTs添加量为10%(w)左右出现拐点,表明此时MWCNTs形成了连续网络,而后电导率趋于稳定。在电导率稳定的区域,PST由于含有噻吩基团,PST/MWCNTs电导率明显高于PS/MWCNTs,且PST4/MWCNTs,PST5/MWCNTs,PT/MWCNTs的电导率比PS/MWCNTs高出一个数量级,这表明共聚物侧基的噻吩含量增加可使导电通路更易形成,从而使材料电导率提高。PST-g-T/MWCNTs的电导率进一步提高。其中,PST2-g-T/MWCNTs和PST3-g-T/MWCNTs的电导率均高于相应的未接枝试样。对比噻吩含量相近的PT(36.1%)/MWCNTs和PST3-g-T(37.8%)/MWCNTs发现,二者的电导率相近,说明通过延长噻吩侧基长度也可达到相近效果;而两种聚合物的Tg分别为23.3℃和86.3 ℃,可见通过调控聚合物分子结构能够实现对材料基本物性的设计。PT接枝物掺杂5%(w)的MWCNTs的电导率即可达14 S/m;掺杂20%(w)的MWCNTs时,电导率达690 S/m,而相同掺杂量的PS/MWCNTs电导率仅为17 S/m。

图7 聚合物/MWCNTs复合材料的电导率Fig.7 Conductivity of polymer/multi-walled carbon nanotubes(MWCNTs) composites.
3 结论
1)通过自由基聚合制备了噻吩功能化聚苯乙烯PST,通过调控共聚单体配比可以实现聚合物中的噻吩含量的调节;还可通过对聚合物中的噻吩基进行溴化反应和与2-硼酸噻吩的Suzuki偶联反应,进一步在PST上接枝噻吩基。
2)溴化反应和噻吩接枝反应均可高效完成,通过逐步接枝噻吩基的方法,能够对PST的组成、热性能和加工性能进行逐步调控,使PST保持可溶解、可加工的优点。
3)利用PST与MWCNTs制备复合材料,该材料的电导率显著高于PS/MWCNTs复合材料,且材料的电导率随聚合物中噻吩含量增加而提高。
猜你喜欢
橡胶科技(2022年9期)2022-12-12 05:26:53
合成树脂及塑料(2020年6期)2020-12-29 07:02:02
石油沥青(2019年4期)2019-09-02 01:41:54
潍坊学院学报(2017年2期)2017-04-20 08:44:22
中国塑料(2016年3期)2016-06-15 20:30:03
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
中国塑料(2015年1期)2015-10-14 00:58:41
橡胶工业(2015年9期)2015-08-29 06:40:46
应用化工(2014年9期)2014-08-10 14:05:08
