
液相色谱-串联质谱法测定植物源性食品中总氟吡禾灵残留
2021-11-03 09:17王兴宁唐亚峰梁艺馨
分析测试学报 2021年10期
王兴宁,王 涛,李 洁,李 志,唐亚峰,梁艺馨,朱 明
(1. 贵阳海关综合技术中心,贵州 贵阳 550081;2. 贵州轻工职业技术学院,贵州 贵阳 540003)
氟吡禾灵属于苯氧羧酸类除草剂,主要以氟吡甲禾灵和氟吡乙禾灵2 种形式生产。两者均为出苗前和出苗后选择性除草剂,用于控制阔叶作物中的禾本科杂草,其机理是抑制乙酰辅酶A 羧化酶(ACCase),导致脂肪酸合成受阻[1-2]。该化合物的分子结构含有酯和羧基,其酯类在土壤或植物中迅速降解为其母体酸,并通过酯键、糖苷键或其他键与基质组分共价结合,在作物中形成二级共轭残基[3-4]。苯氧羧酸类除草剂及其代谢降解产物对哺乳动物、鱼类、鸟类和人类毒性很低,但摄入一定量的该类除草剂可引起腹泻、食欲减退、抑郁,出现肺、肝脏、脾脏和脑膜的严重充血等毒性反应。因此,欧盟、日本和美国等国家和地区均对其在农产品中的残留规定了严格限量。
GB 2763-2019《食品安全国家标准食品中农药最大残留限量》规定了氟吡甲禾灵、氟吡禾灵及其共轭物之和,以氟吡甲禾灵表示,在葵花籽、结球甘蓝及柑橘等植物源性食品中的最大残留限量为0.02~3 mg/kg[5]。欧盟农药残留法规(EU)No.396/2005 修订了植物源性食品中氟吡禾灵(总氟吡禾灵,氟吡禾灵及其酯、盐和共轭物的总和,以氟吡禾灵表示)的最大残留限量为0.01~0.5 mg/kg[6]。日本肯定列表规定氟吡禾灵在豌豆、洋葱及柑橘等植物源性食品中的最大残留限量为0.01~0.5 mg/kg[7]。然而,当前国内外对植物源性食品中总氟吡禾灵的测定方法标准甚少。文献报道氟吡禾灵的检测方法主要有气相色谱法[8-9]、气相色谱-质谱法[10]、液相色谱法[11]和液相色谱-串联质谱法[12-18]等。运用气相色谱法和气相色谱-质谱法时,需对目标物进行衍生化,存在衍生副产物干扰严重、回收率低等缺点;液相色谱法的灵敏度高,但在检测复杂基质(如茶叶样品)时确证困难;液相色谱-串联质谱法是检测氟吡禾灵的最佳技术手段,但目前的研究以检测单一目标物的氟吡禾灵和氟吡甲禾灵残留为主,对总氟吡禾灵残留缺少较为全面系统的研究,同时亦未对大米、柠檬、柑橘、马铃薯、茶叶等9 种植物源性食品基质进行考察。本文采用基质分散固相萃取进行样品前处理,结合液相色谱-串联质谱建立了植物源性食品中总氟吡禾灵残留的测定方法。实验优化了各种基质样品的最佳水解条件,以评估氟吡禾灵酯和共轭物的转化效率,同时优化了基质分散固相萃取条件,以适应各种基质类型样品的净化。该方法可为总氟吡禾灵残留日常监控提供实用的技术手段。
1 实验部分
1.1 仪器与试剂
Sciex API 5500 Q型超高效液相色谱-串联质谱仪,配备电喷雾离子源和Analyst1.5.1工作站(美国AB公司),高速分散机(i国IKA公司),Sigma 3K15离心机(美国Sigma公司)。
乙腈、甲醇、乙酸铵、甲酸(色谱纯,上海安谱有限公司);实验用水为Elix/Milli-Q 高纯水(美国Millipore 公司);氢氧化钠、硫酸(优级纯,上海国药集团);无水硫酸镁、乙酸钠(农残级,阿拉丁试剂公司);N-丙基乙二胺(PSA,40~60 μm)、石墨化炭黑(GCB,30~90 μm)、十八烷基硅烷(C18,50 μm,60 Å)(农残级,上海安谱有限公司)。
氟吡禾灵(CAS 号69806-34-4)、氟吡甲禾灵(CAS 号69806-40-2)和氟吡乙禾灵(CAS 号87237-48-7)(北京坛墨质量检测技术有限公司);同位素内标(±)氟吡禾灵-D4(纯度99.0%,加拿大Toronto Research Chemical公司)。
以乙腈为溶剂,分别配制质量浓度为100 μg/mL 的氟吡禾灵、氟吡甲禾灵、氟吡乙禾灵和氟吡禾灵-D4(内标)的单个标准储备溶液,并于-20 ℃下储存。以乙腈为溶剂,配制含有氟吡禾灵、氟吡甲禾灵及氟吡乙禾灵2.5、5.0、10、25、50 ng/mL(含内标50 ng/mL)的系列混合标准溶液,现配现用。
植物源性食品(柠檬、柑橘、马铃薯、结球甘蓝、甜椒、洋葱、大米、葵花籽及茶叶)的样品购自贵阳当地超市。
1.2 仪器条件
1.2.1 色谱条件 ACQUITYUPLC BEH C18色谱柱(2.1 mm×50 mm,1.7 μm,Waters 公司);柱温为40 ℃;样品室温度为15 ℃;进样量为10 μL;流动相:A为0.1%甲酸水溶液,B为0.1%甲酸乙腈;流速为0.2 mL/min。梯度洗脱程序如下:0~0.20 min,10% B;0.20~3.00 min,10%~50% B;3.00~7.00 min,50%~75%B;7.00~7.01 min,75%~10%B;7.01~10.0 min,10%B。
1.2.2 质谱条件 电喷雾离子源(ESI);多反应监测(MRM);正离子扫描;离子化电压为-4 500 V;离子源温度为400 ℃;气帘气压力为1.38×105Pa;离子源喷雾气压力为2.07×105Pa;碰撞气压力为4.14×104Pa。MRM模式下的质谱参数见表1。
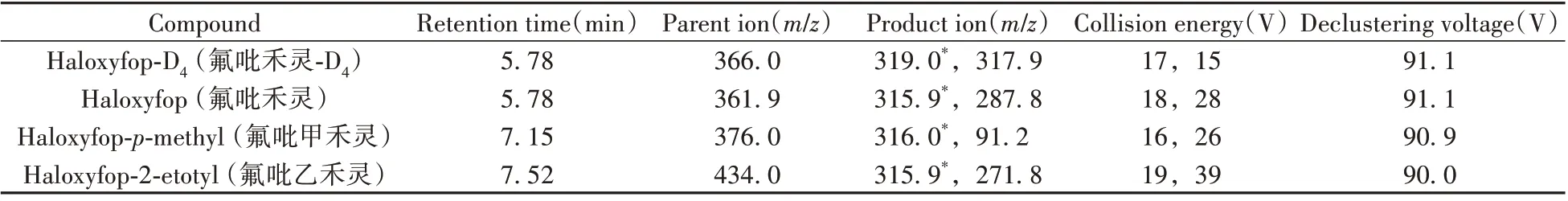
表1 MRM模式下的质谱参数Table 1 MS parameters in MRM mode
1.3 样品提取与净化
1.3.1 高水分含量样品的提取 准确称取5 g 样品(精确至0.01 g)置于50 mL 聚丙烯离心管中,加入50 μL 内标(10 μg/mL)涡旋混匀,再加入10 mL 乙腈和2 mL 5 mol/L NaOH 涡旋混匀,40 ℃水浴超声水解和提取30 min,加入2 mL 2.5 mol/L H2SO4涡旋混合,酸化后的样品提取液pH 值为5~7,加入6 g无水MgSO4和1.5 g乙酸钠剧烈振摇30 s,以5 000 r/min离心5 min,收集上清液。
1.3.2 低水分含量样品的提取 准确称取5 g 样品(精确至0.01 g)置于50 mL 聚丙烯离心管中,茶叶样品称取2 g(精确至0.01 g),加入50 μL 内标(10 μg/mL)涡旋混匀,加入10 mL 水涡旋混合后静置30 min,再加入10 mL 乙腈和2 mL 5 mol/L NaOH涡旋混匀,后续操作同“1.3.1”。
1.3.3 净 化 在2 mL微型离心管中预装150 mg无水MgSO4、50 mg C18和50 mg GCB,吸取提取的乙腈相至离心管中,涡旋振荡5 min 后6 000 r/min 离心1 min,上清液经0.22 μm 有机滤膜过滤于2 mL 进样小瓶,待测定。
2 结果与讨论
2.1 质谱条件的优化
比较了ESI-和ESI+两种模式下氟吡禾灵、氟吡禾灵-D4、氟吡甲禾灵和氟吡乙禾灵的信号强度。以含5 mmol/L 乙酸铵的甲醇-水(1∶1,体积比)配制100 ng/mL 上述4种化合物的标准溶液,并以10 mL/min的流速直接注入质谱。结果表明,氟吡禾灵和氟吡禾灵-D4在ESI-和ESI+两种模式下均有较高的信号强度,而氟吡甲禾灵和氟吡乙禾灵仅在ESI+模式下具有较高的信号强度。因此,采用ESI+模式可实现所有目标物的同时监测,并进一步优化ESI+模式下MRM 的母离子、子离子、碰撞能量和去簇电压,每个目标物选择丰度较高、干扰最少的2组离子对用于MRM监测。最终优化的质谱条件如“1.2.2”所示。
2.2 色谱条件的优化
氟吡禾灵和氟吡禾灵酯属于中等极性和弱极性化合物,在弱极性的C18柱上均具有较好的保留。实验比较了Waters ACQUITY UPLC BEH C18(50 mm×2.1 mm,1.7 μm)和Agilent ZORBAX SB-C18(150 mm×4.6 mm,4.6 μm)的分离效果,结果表明,两柱均能较好地分离氟吡禾灵、氟吡甲禾灵和氟吡乙禾灵,前者的分离效果更优,因此选择ACQUITY UPLC BEH C18柱(50 mm×2.1 mm,1.7 μm)。实验分别以甲醇-水和乙腈-水为流动相进行分离,均不能得到良好的峰形,考虑到流动相添加剂(如乙酸铵和甲酸)可显著改善色谱峰的分离度与灵敏度,进一步比较了甲醇-5 mmol/L 乙酸铵水溶液和0.1%甲酸乙腈-0.1%甲酸水溶液的分离效果。结果表明,以0.1%甲酸乙腈-0.1%甲酸水溶液为流动相可获得最佳的分离度和灵敏度。在“1.2”的最佳条件下,目标物的总离子流色谱图见图1。由图可知,目标物的响应强度高、无干扰,分离度高、峰形良好。
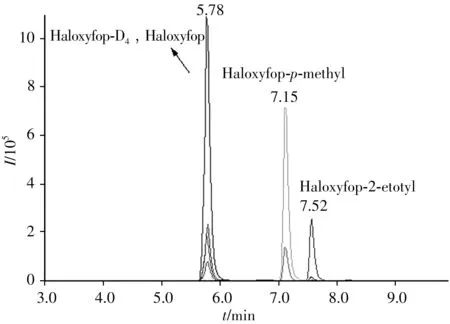
图1 10 ng/mL混合标准工作溶液的总离子流色谱图Fig.1 Chromatogram for mixed standard solution at concentration of 10 ng/mL haloxyfop-D4:50 ng/mL
2.3 提取条件的优化
考察了不同提取方法对复杂基质茶叶样品的去除杂质效果以及提取和水解效果。通过向茶叶样品中添加氟吡甲禾灵和氟吡乙禾灵的标准溶液评估总氟吡禾灵的转化率。比较了以下4种常用的水解和萃取条件:①室温涡旋30 min;②室温涡旋30 min,40 ℃水浴恒温30 min;③40 ℃恒温振荡30 min;④40 ℃超声水解30 min。每个实验均加入1 mL 5 mol/L NaOH进行水解和1.5 mL 2.5 mol/L H2SO4进行中和[14]。结果表明,采用方法④获得的总氟吡禾灵转化率最高,因此选择样品提取条件为40 ℃超声水解30 min。
2.4 水解条件的选择
氟吡禾灵酯类主要有氟吡甲禾灵和氟吡乙禾灵,在施用农作物后的短时间内,所有酯类均降解为氟吡禾灵[4]。当前采用碱性水解等方式将氟吡禾灵酯类、盐及共轭物直接转化为氟吡禾灵,测定氟吡禾灵总和,与欧盟农残限量规定表示一致,并以氟吡禾灵酯类的转化情况作为评价标准[13]。碱性条件有助于提高氟吡禾灵的转化率,但过量的碱可能导致目标物分解和随后的中和困难。选择柠檬样品(酸性样品消耗最多的碱)添加0.01 mg/kg 的氟吡甲禾灵和氟吡乙禾灵标准溶液,考察了5 mol/L NaOH 用量(0.5、1.0、1.5、2.0、2.5 mL)对总氟吡禾灵转化率的影响(图2)。结果表明,5 mol/L NaOH 用量为2.0 mL和2.5 mL时总氟吡禾灵的转化率较高并趋于平稳。实验最终确定5 mol/L NaOH 用量为2 mL,可适用于大多数类型样品基质的水解。
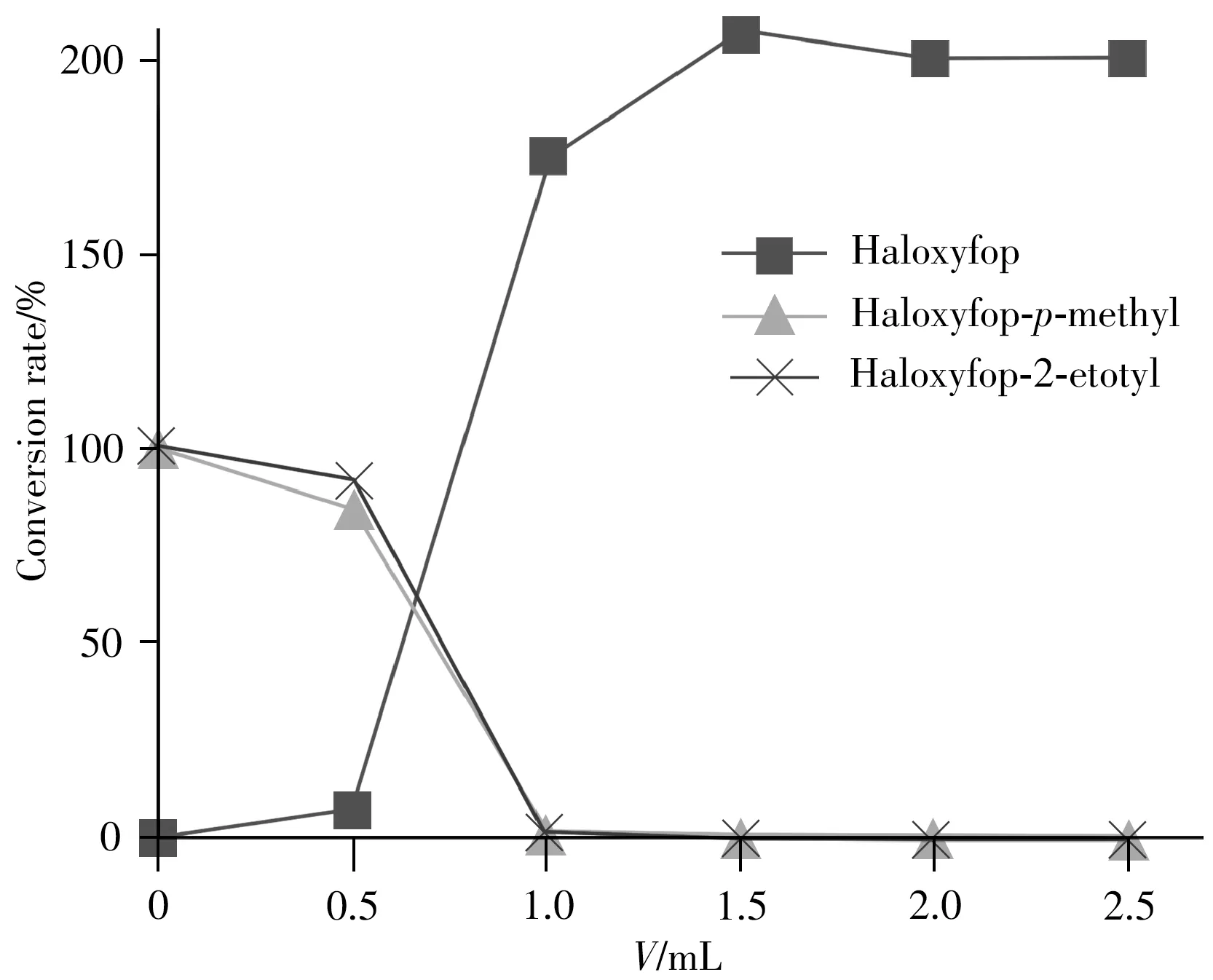
图2 不同体积碱用量对柠檬样品中总氟吡禾灵的水解效果Fig.2 Hydrolysis efficiency for total haloxyfop with different volume alkali in lemon sample spiked at 0.01 mg/kg haloxyfop-p-methyl and haloxyfop-2-etotyl
2.5 净化方法的优化
基质分散固相萃取具有快速、操作简单、吸附能力强等特点,被广泛用于食品中农药残留检测[19]。按照优化的提取方法对添加0.01 mg/kg 氟吡禾灵标准溶液的茶叶样品进行处理,提取液转入2 mL 预装有4 种不同比例吸附剂(MgSO4、C18、PSA和GCB)的微型离心管中进行净化(图3)。不同吸附剂的组合如下:①MgSO4150 mg 和C1825 mg;②MgSO4150 mg 和C1850 mg;③MgSO4150 mg和C18100 mg;④MgSO4150 mg、C1825 mg 和PSA 25 mg;⑤MgSO4150 mg、C1825 mg 和GCB 25 mg;⑥MgSO4150 mg、C1825 mg、PSA 25 mg 和GCB 25 mg。结果表明,组合①、②、③和⑤均可获得满意的回收率,而由于PSA 对目标物具有吸附作用,导致组合④和⑥的回收率下降。考虑植物源性食品基质的复杂性,最终确定适用于广泛基质范围的吸附剂组合MgSO4150 mg、C1850 mg和GCB 50 mg。
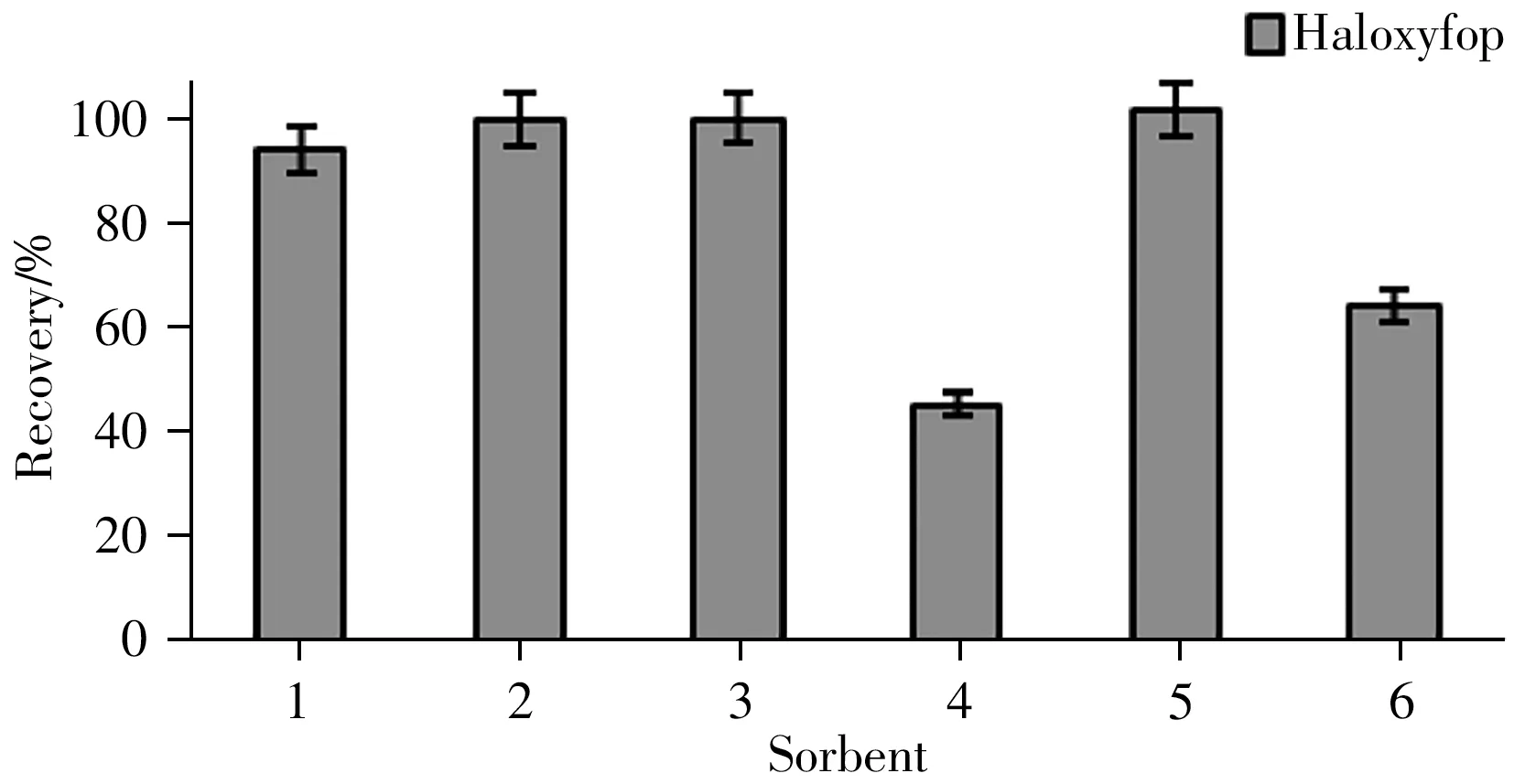
图3 不同吸附剂组合对茶叶样品(添加0.01 mg/kg氟吡禾灵)的回收率Fig.3 Recovery of tea samples clean-up with different sorbents(spiked at 0.01 mg/kg haloxyfop)
2.6 基质效应
植物源性食品中色素、有机酸和脂肪等杂质易引起基质增强或抑制效应,研究表明基质种类、基质数量和目标物化学性质对基质效应有一定影响[20]。实验采用不同基质代表性样品,如茶叶(复杂基质样品)、柠檬(高水和高酸样品)和大米(低水和高淀粉样品)制备基质匹配标准曲线。按照下式计算基质效应(ME):ME=(m基质-m溶剂)/m溶剂×100%,其中m基质和m溶剂分别为基质和溶剂匹配标准曲线的斜率。结果表明,氟吡禾灵在茶叶、柠檬和大米中的基质效应较小,ME为-9.2%~-4.5%,ME低于20%其影响可忽略[21]。因此,本方法采用溶剂匹配标准曲线进行定量。
2.7 稳定性
通过改变碱水解与酸中和的参数评估该方法的稳定性[21]。用添加0.01 mg/kg 氟吡甲禾灵的茶叶样品进行考察,碱水解时,将5 mol/L NaOH 的体积由2 mL改变为1.5 mL;酸中和时,将2.5 mol/L H2SO4的体积由2 mL(溶液的pH ≈4~5)改变为1.5 mL(溶液的pH ≈6~7)。结果表明,改变碱水解参数前获得的平均回收率为94.3%,相对标准偏差(RSD,n=5)为5.6%,改变后获得的平均回收率降至87.2%(RSD 为4.2%,n=5);改变酸中和参数前获得的平均回收率为94.5%(RSD 为2.5%,n=5),改变后获得的平均回收率为98.3%(RSD为7.3%,n=5)。结果表明,碱水解5 mol/L NaOH 体积的改变对方法稳健性的影响显著。因此在实验过程中应严格控制,以避免结果的偏离。
2.8 方法学考察
2.8.1 线性范围 通过内标法(氟吡禾灵-D4作为内标,质量浓度为50 ng/mL)配制2.5、5.0、10、25、50 ng/mL的系列基质匹配和溶剂匹配标准工作溶液进行LC-MS/MS分析。以质量浓度为横坐标(x,μg/L),分析物与内标的峰面积比(y)为纵坐标绘制标准曲线。结果表明,氟吡禾灵在2.5~50 ng/mL 范围内线性关系良好,相关系数(r2)大于0.999。
2.8.2 回收率与相对标准偏差 取空白样品,分别添加氟吡禾灵、氟吡甲禾灵和氟吡乙禾灵标准溶液,加标水平为0.01、0.05、0.1 mg/kg,每个水平重复5次,并根据氟吡禾灵的标准曲线以及氟吡甲禾灵和氟吡乙禾灵水解为氟吡禾灵的转化系数(分子量比分别为1.04和1.20)计算最终的回收率结果。本方法同时还能监测氟吡甲禾灵和氟吡乙禾灵水解后的残留情况。结果表明,柠檬、柑橘、马铃薯、结球甘蓝、甜椒、洋葱、大米、葵花籽及茶叶9种基质的平均回收率为80.7%~114%,RSD为1.4%~11%(见表2)。
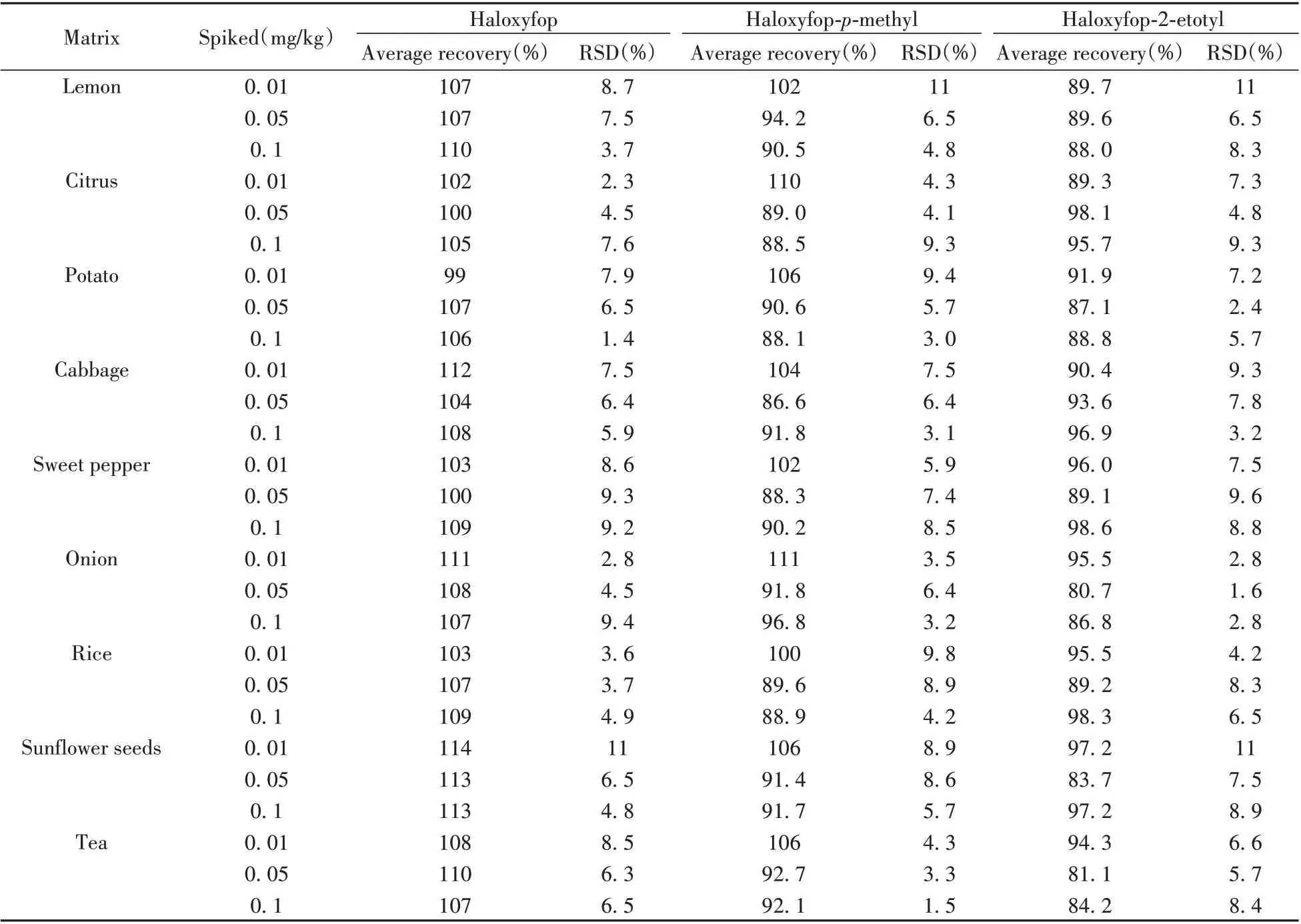
表2 空白样品的平均回收率及相对标准偏差(n=5)Table 2 Mean recoveries and RSDs in blank samples(n=5)
2.8.3 定量下限 在欧盟(EU)No.396/2005法规[6]中,大多数植物源性食品中总氟吡禾灵的最大允许残留限量(MRL)低至0.01 mg/kg。本方法的定量下限(LOQ)采用加标回收方法进行验证,柠檬、柑橘、马铃薯、大米、茶叶等9 种样品的LOQ 均能达到0.01 mg/kg,满足国内外法规对总氟吡禾灵残留限量的检测要求。
2.9 实际样品的检测
应用本方法检测柠檬、柑橘、马铃薯、结球甘蓝、甜椒、洋葱、大米、葵花籽、茶叶及小麦等样品中的总氟吡禾灵残留,结果在2批次的小麦粉样品中检出总氟吡禾灵,残留量分别为0.03、0.12 mg/kg,其余样品均未检出。
3 结 论
本研究建立了液相色谱-串联质谱快速分析植物源性食品中总氟吡禾灵的方法。优化的前处理步骤可将氟吡禾灵酯类完全转化为总氟吡禾灵;方法的线性关系良好,r2大于0.999;柠檬、柑橘、马铃薯、结球甘蓝、甜椒、洋葱、大米、葵花籽及茶叶9 种样品的加标回收率为80.7%~114%,RSD 为1.4%~11%;定量下限低至0.01 mg/kg,可满足国内外农药残留限量法规的要求。该方法具有灵敏度高、稳定性好、专一性强、准确度高的优势,为总氟吡禾灵残留的日常监控检测提供了实用的技术手段。
猜你喜欢
康复(2022年25期)2022-11-18
基层中医药(2022年7期)2022-11-17
现代仪器与医疗(2022年2期)2022-08-11
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
中学化学(2019年3期)2019-07-08
中学化学(2016年2期)2016-05-31
课程教育研究·下(2016年2期)2016-03-25
中国民族民间医药·下半月(2014年2期)2014-09-26
中国信息化·学术版(2013年3期)2013-06-25
