
磺苄西林钠有关物质研究
2021-11-01 05:33彭琳艾俊涛廖彬刘雁鸣湖南省医疗器械检验检测所长沙4004湖南省药品检验研究院长沙400
中南药学 2021年10期
彭琳,艾俊涛*,廖彬,刘雁鸣(. 湖南省医疗器械检验检测所,长沙 4004;. 湖南省药品检验研究院,长沙400)
磺苄西林钠(sulbenicillin sodium)为半合成广谱青霉素类抗菌药物,应用于铜绿假单胞菌、肠杆菌属、变形菌和其他敏感菌所致的系统感染[1-3];1972年由日本武田药品公司研究所研发,20世纪80年代在我国开始使用;《中国药典》于1990年版起收录本品。现行质量标准收载于《中国药典》2020年版二部[4]。标准中采用HPLC自身对照法测定有关物质,要求单个杂质峰面积不得大于对照溶液主峰与相对保留时间0.9处的色谱峰面积和的2倍(2.0%),各杂质峰面积的和不得大于对照溶液主峰相对保留时间0.9处的色谱峰面积和的4倍(4.0%),供试品溶液色谱图中小于对照溶液主峰与相对保留时间0.9处的色谱峰面积和的0.05倍的峰忽略不计。标准中未规定杂质的具体种类。分析磺苄西林钠的合成路线(见图1)[5-7],其采用的主要原料有:6-氨基青霉烷酸(6-APA)、磺苯乙酰氯,合成工艺见图1。本研究对合成工艺过程中可能引入的多种特定杂质进行方法学验证及定量研究,以用于本品质量控制和工艺稳定性考察。

图1 磺苄西林钠合成工艺路线图Fig 1 Synthesis route of sulbenicillin sodium
1 仪器与试药
LC-30A岛津高效液相色谱仪,SPD-M20A二极管阵列检测器(岛津公司),Labsolution色谱数据工作站(岛津公司),NewClassic电子天平(Mettler toledo公司,精度:0.1 mg);磷酸二氢钾(分析纯,国药集团化学试剂有限公司),甲醇(色谱纯,TEDIA公司)。对照品信息见表1。自制样品[编号1、2、3,含量:每1 mg的效价为1140磺苄西林单位(抗生素微生物检定法)],对照样品[编号4,来源:某制药企业已上市产品,含量:每1 mg的效价为1140磺苄西林单位(抗生素微生物检定法)]。

表1 对照品信息Tab 1 Reference information
2 方法与结果
2.1 溶液的制备
2.1.1 供试品溶液 取样品,精密称定,加流动相溶解并稀释制成每1 mL中约含0.2 mg的溶液。
2.1.2 对照溶液 精密量取上述供试品溶液1 mL,置50 mL量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.3 混合对照品储备液 分别取6-APA、磺苯乙酰氯及左旋磺苯乙酸适量,加流动相溶解并定量稀释制成每1 mL含以上杂质约为10 μg的混合对照品储备液。
2.1.4 线性溶液 分别精密量取“2.1.3”项下混合对照品储备液适量,加流动相定量稀释,配成系列标准工作溶液,作为线性测定溶液。
2.1.5 破坏试验用溶液
①酸破坏:取样品(编号1)10 mg,精密称定,置50 mL量瓶中,加0.05 mol·L-1盐酸溶液1.5 mL溶解,放置0.5 h,用0.05 mol·L-1氢氧化钠溶液中和,用流动相稀释至刻度,作为酸破坏供试品溶液。
②碱破坏:取样品(编号1)10 mg,精密称定,置50 mL量瓶中,加0.1 mol·L-1氢氧化钠溶液1.5 mL溶解,放置2 min,用0.1 mol·L-1盐酸溶液中和,用流动相稀释至刻度,作为碱破坏供试品溶液。
③氧化破坏:取样品(编号1)10 mg,精密称定,置50 mL量瓶中,加流动相5 mL溶解后,加30%过氧化氢溶液2滴,放置0.5 h,用流动相稀释至刻度,作为氧化破坏供试品溶液。
④热破坏:取在120℃烤箱中加热3 h后冷却至室温的样品(编号1),用流动相溶解并稀释至刻度,作为热破坏供试品溶液。
⑤光破坏:取样品(编号1),在光强度4500 Lx下照射 48 h后,稀释剂稀释至刻度,作为光破坏供试品溶液。
2.2 色谱方法
磺苄西林钠质量标准收载于《中国药典》2020年版二部及JP17,某制药公司生产的本品执行标准为国家食品药品监督管理局标准YBH06232010。各标准中有关物质方法存在以下差别,见表2。

表2 各质量标准有关物质方法比较Tab 2 Related substance in different standards
本次自制产品为技术转化,合成工艺完全与该制药公司合成工艺一致,未作修改,故按照YBH06232010标准方法对有关物质方法进行验证:色谱柱为Acclaim 120 C18(4.6 mm×250 mm,5 µm);检测波长:230 nm;流速:1.0 mL·min-1;柱温:35℃;进样体积:20 μL;流动相:0.02 mol·L-1磷酸二氢钾溶液-甲醇(80∶20)。
2.3 专属性试验
精密量取流动相20 μL,进样测定,结果溶剂空白仅在2 min左右出现溶剂峰,但在主峰与杂质峰位置处不显示色谱峰,结果表明空白溶剂不干扰有关物质测定,见图2A。
2.4 破坏试验
按照“2.2”项下色谱条件,分别测定“2.1.5”项下的不同酸、碱、氧化、热与光破坏条件下制备的各种相应供试品溶液,结果分别与未经破坏的样品进行比较,分析杂质谱的变化情况,结果见图2,样品杂质情况见表3。
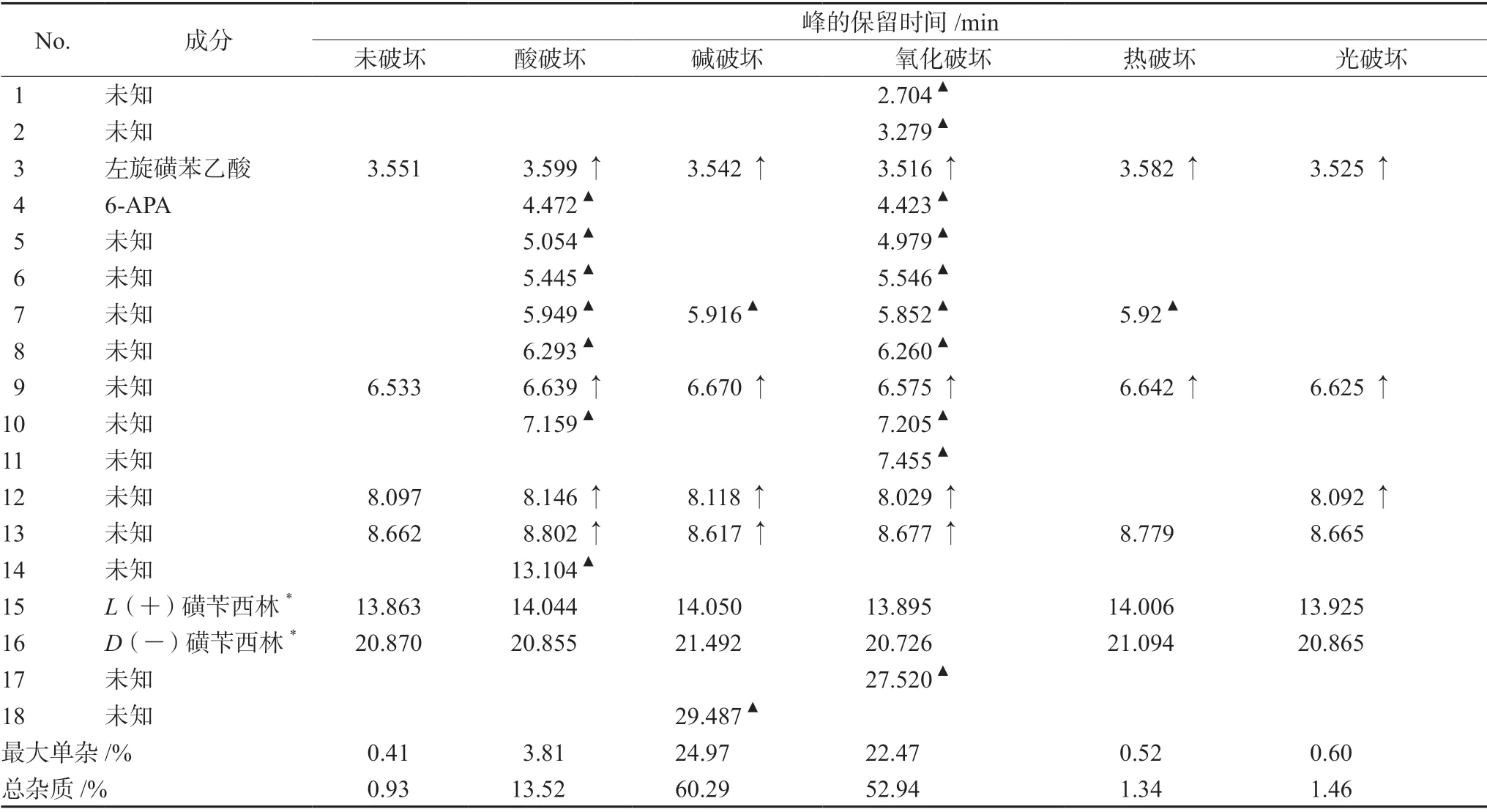
表3 破坏试验杂质谱Tab 3 Impurity profiling of destruction test
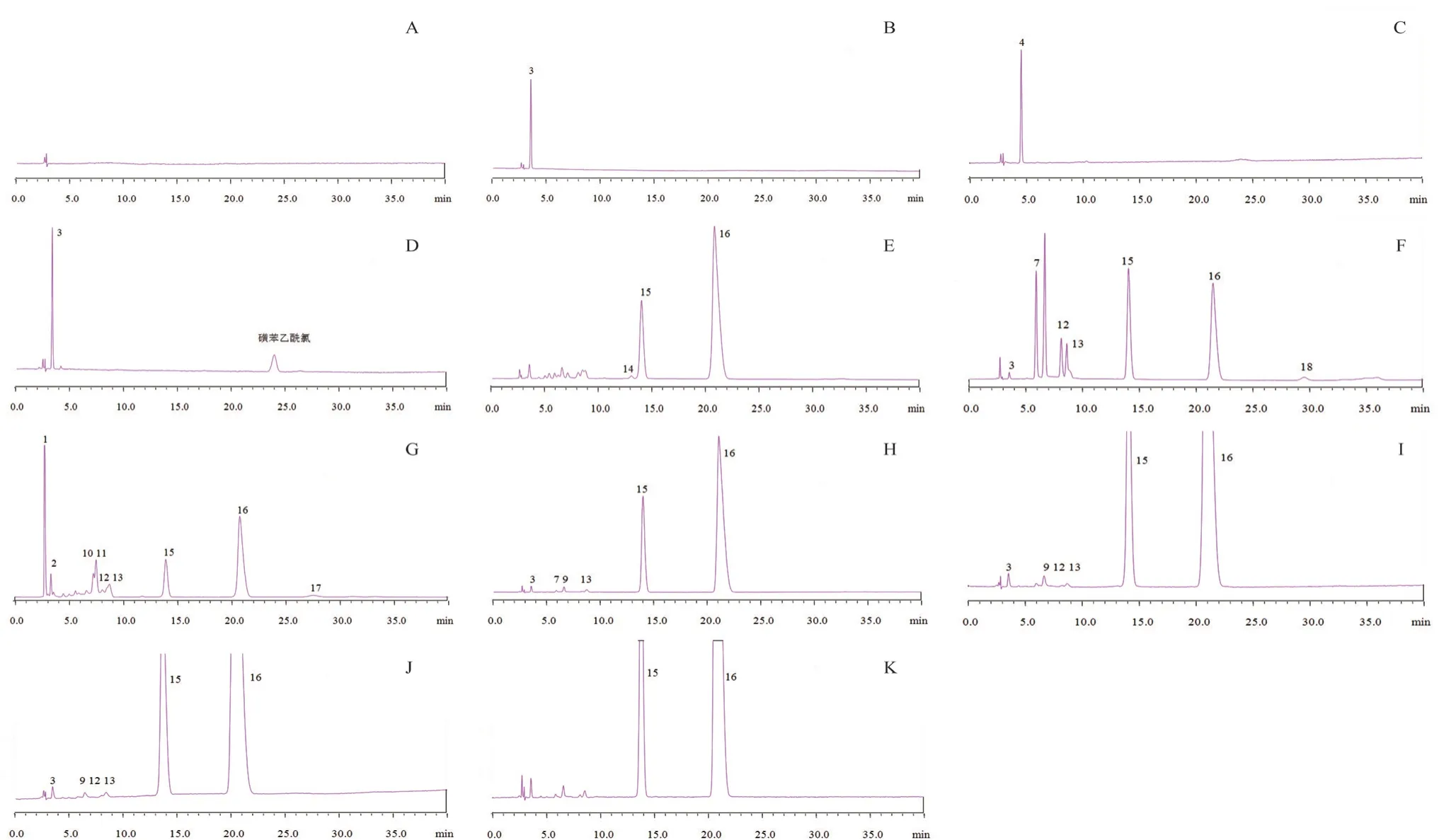
图2 专属性实验色谱图Fig 2 Chromatograms of specificity test
结果,①酸破坏:经酸破坏0.5 h出现多个未知的明显的杂质,主峰下降11.1%。②碱破坏:经碱破坏2 min即出现多个未知的明显的杂质,主峰下降39.7%;破坏1.5 h主成分已被完全破坏。③氧化破坏:样品对氧化剂不稳定,经氧化破坏0.5 h出现多个未知的明显的杂质,主峰下降50.2%。④热破坏和光破坏使样品的总杂质略为增加。在该色谱条件下,样品经酸、碱、氧化、高温、光降解所产生的降解产物均能与主峰完全分离,该系统适合检测本品有关物质。同时说明样品具备一定的热和光稳定性,但在酸、碱、氧化条件下均不稳定。
2.5 杂质线性关系考察
按“2.1”项下的制备方法和“2.2”项下色谱条件,分别测定6-APA、左旋磺苯乙酸及磺苯乙酰氯的系列标准工作溶液,记录色谱图,以质量浓度为x轴,峰面积为y轴,进行线性回归,结果6-APA回归方程为:y=7688.3x-208.87,r=1.000;左旋磺苯乙酸回归方程为:y=4118.9x+448.33,r=0.9998;磺苯乙酰氯回归方程为:y=1892.6x+673.78,r=0.9991。结 果6-APA在1.113~11.130 μg·mL-1、左旋磺苯乙酸在1.217~12.170 μg·mL-1与峰面积呈良好的线性关系;磺苯乙酰氯在1.284~12.840μg·mL-1与峰面积呈良好的线性关系。
2.6 检测限与定量限
取各杂质线性最低点,对其色谱图中数据计算方法检测限与定量限,结果6-APA、左旋磺苯乙酸、磺苯乙酰氯的检测限分别为0.009、0.014、0.035 μg,定量限分别为0.029、0.046、0.116 μg。
2.7 回收试验
取样品(编号1)约20 mg,精密称定,置10 mL量瓶中,共9份,加流动相溶解并稀释至刻度,各分别取1 mL置10 mL量瓶中,分别精密加入混合对照品储备液4.8、4.0、3.2 mL,各3份,加流动相稀释并定容至刻度,作为回收供试品溶液,测定并计算回收率。结果6-APA高、中、低浓度的平均回收率分别为94.5%、93.1%、94.3%,RSD分别为0.82%、0.63%、0.83%(n=3);左旋磺苯乙酸高、中、低浓度的平均回收率分别为101.4%、102.4%、102.2%,RSD分别为0.42%、0.84%、1.1%(n=3);磺苯乙酰氯高、中、低浓度的平均回收率为102.2%、101.9%、103.4%,RSD分别为1.1%、1.2%、1.8%(n=3),说明本法准确可行。
2.8 稳定性考察
按照“2.2”项下色谱条件,分别在0、2、4、8、16、24 h测定同一供试品溶液,以杂质含量为考察指标,结果最大单杂及总杂质分别在0.41%~0.46%、0.89%~1.01%,相应的RSD分别为5.3%、5.1%,说明溶液在24 h内稳定。
2.9 重复性试验
按照 “2.2”项下色谱条件,连续测定6份供试品溶液及对照品溶液,以杂质含量为考察指标,结果最大单杂及总杂质的平均含量分别为0.43%、0.89%,RSD分别为3.8%、2.0%,说明方法重复性结果好。
2.10 耐用性试验
取样品(编号1),按“2.1”项下方法制备,分别考察流动相比例变化20%、检测波长变化±5 nm、柱温变化±5℃、流速变化20%及不同品牌的色谱柱,结果见表4。以上结果表明变换波长、柱温、流速、流动相比例及色谱柱,测试结果变化均在很小的范围内,说明上述5种条件的微小变化对测试结果无影响。
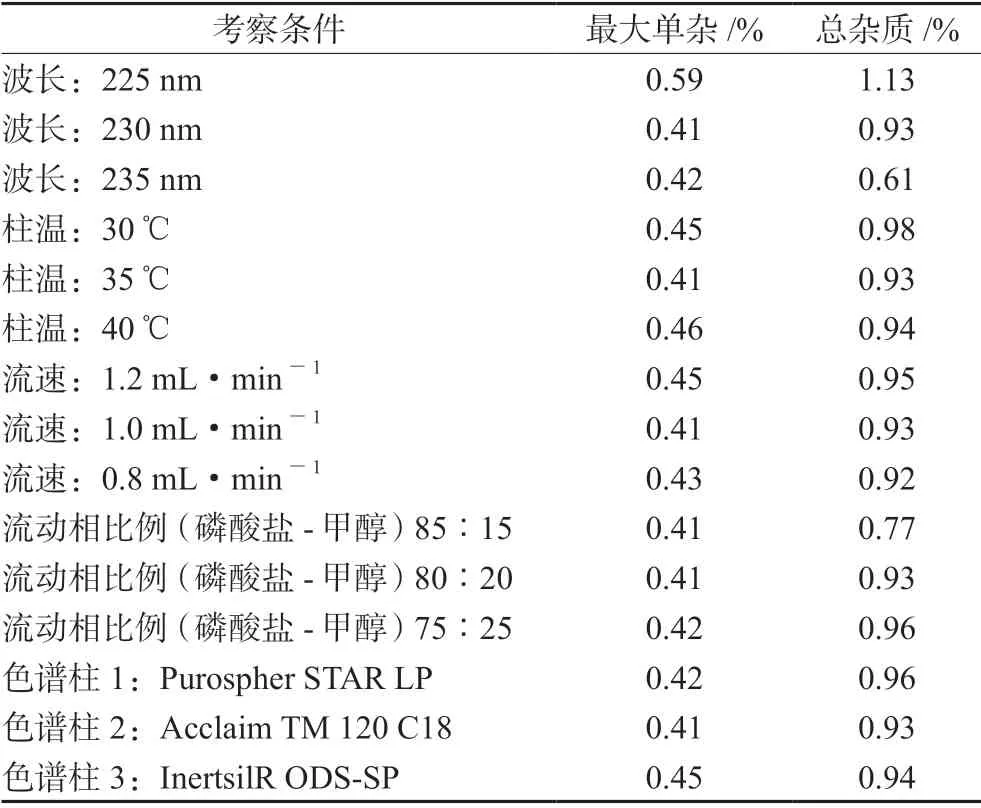
表4 耐用性试验结果Tab 4 Different condition
2.11 样品测定
按“2.1”项下的制备方法和“2.2”项下色谱条件,测定自制3批样品及1批对照样品,列表比较自制样品与对照样品杂质检出情况,结果见表5。自制样品杂质谱与对照样品的杂质谱略有差别,对照样品杂质峰的数量比自制样品多1个,自制样品最大单杂及总杂质的量与对照样品相当。

表5 杂质谱对比Tab 5 Impurity profile
3 讨论
3.1 色谱方法的选择
磺苄西林钠质量标准收载于《中国药典》2020年版二部及JP17,各质量标准有关物质方法比较见表2。本研究按照某制药公司批准的执行标准YBH06232010的有关物质测定方法,该方法在流动相的组成、检测波长、色谱图记录时长、限度要求等方面与药典标准略有差异。因本次自制产品为技术转化,合成工艺与该制药公司一致,未作修改,故按照YBH06232010标准方法对有关物质方法进行验证,以便于自制产品与原研产品测定结果的对比分析。
同时,《中国药典》2015年版二部中规定记录色谱图至主成分峰保留时间的4.5倍,本研究给出的色谱图显示记录至40 min,未达到4.5倍,在方法建立过程中已进行过考察,能确保将所有杂质成分都能洗脱出来,故仍按照YBH06232010方法,采用检测时间40 min进行验证。
3.2 有关物质特定杂质的控制
磺苄西林钠的现行质量标准《中国药典》2015年版二部中对单杂及总杂质量有控制,未对具体的单个杂质量进行限量规定。根据本产品的有关物质考察结果,成品中含有未反应完全的起始物料6-APA,经一定程度破坏还会产生其他杂质,如左旋磺苯乙酸(原料磺苯乙酰氯带入),因此建议根据产品的工艺特性增加特定杂质的限度要求。
3.3 破坏试验结果分析
强制降解试验显示,磺苄西林钠在氧化破坏及酸破坏试验中产生的杂质较多,根据青霉素类抗菌药物降解反应的特点[8],推测其主要是β-内酰胺抗菌药物的水解产物,其具体杂质结构有待于进一步研究。研究提示本品与酸性药物存在配伍禁忌,且易被氧化分解,同时热和光破坏也会使杂质量略微增加,因此建议本品的储存、运输过程中避光、避热、密封保存。
3.4 小结
综上,有关物质[9-13]是与生产工艺、生产过程控制水平密切相关的项目,对药品安全性影响较大,目前杂质水平已经成为药品质量控制中至关重要的一个环节。运用各项检测技术及知识,建立磺苄西林钠中杂质的测定方法,灵敏度高,分离效果好,可准确定性,为其工艺生产及储存运输提供有效的信息,为磺苄西林钠原料及其注射液质量控制提供一定的参考依据。
猜你喜欢
Chinese Physics B(2022年5期)2022-05-16
艺术品鉴(2020年6期)2020-12-06
东坡赤壁诗词(2020年3期)2020-07-04
山西中医药大学学报(2020年2期)2020-06-06
启迪与智慧·下旬刊(2019年2期)2019-09-10
中成药(2018年12期)2018-12-29
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
新丝路(下旬)(2016年10期)2016-06-05
Asian Journal of Urology(2015年3期)2015-12-16
