
117例儿童脊髓性肌萎缩症自然病史分析
2021-10-30 02:35:02杨贇滢袁萍李梅蒋莉洪思琦
中国当代儿科杂志 2021年10期
杨贇滢 袁萍 李梅 蒋莉 洪思琦
(重庆医科大学附属儿童医院神经内科/国家儿童健康与疾病临床医学研究中心/儿童发育疾病研究教育部重点实验室/儿科发育重大疾病国家国际科技合作基地/认知发育与学习记忆障碍转化医学重庆市重点实验室,重庆 400014)
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是婴幼儿时期最常见的致死性神经遗传性疾病,是由位于5q11-13的运动神经元存活(survival motor neuron,SMN)基因突变所致,该病为常染色体隐性遗传,人群发病率为1/6 000~1/10 000,人群携带率为1/40~1/60[1]。SMA以进行性运动功能下降为主要表现。近年来随着基因修复、基因替代治疗药物的逐步应用,SMA传统的发展轨迹正在被改变[2]。本研究拟对重庆及周边地区SMA患儿的自然病史进行总结,为进一步开展SMA的综合管理、基因治疗及基因修饰治疗提供临床依据。
1 资料与方法
1.1 研究对象
收集2011年6月至2019年1月我院收治的117例SMA患儿资料。纳入标准:(1)就诊年龄≤18岁者;(2)SMN1基因异常者;(3)未接受基因修饰治疗者;(4)完成随访者。排除标准:(1)临床资料不完整者;(2)生存随访缺失者。所有患儿按照《脊髓性肌萎缩症多学科管理专家共识》[3]的标准进行诊断及分型。
本研究已获得重庆医科大学附属儿童医院伦理委员会批准[(2020)年伦审(研)第(120)号]。
1.2 资料收集
收集SMA患儿的临床资料:(1)流行病学及人口学资料;(2)临床症状及体征;(3)SMN1基因检测结果(采用实时荧光探针定量PCR法);(4)门诊或电话随访生存现状,随访时间截至生命终点或2020年2月。
1.3 统计学分析
采用SPSS 23.0统计学软件进行数据分析。非正态分布的计量资料采用中位数(四分位数间距)[M(IQR)]或中位数(范围)表示,多组间比较采用Kruskal-WallisH检验并进行组间两两比较。计数资料采用例数或百分率(%)表示,多组间比较采用χ2检验或Fisher确切概率法,组间两两比较采用χ2分割法。采用Kaplan-Meier分析1型SMA患儿的生存状态,生存率比较采用log-rank检验;采用Cox比例风险回归模型分析影响1型SMA患儿生存状态的因素。χ2分割法检验水准以P<0.0167为差异有统计学意义,其他检验水准以P<0.05为差异有统计学意义。
2 结果
2.1 一般资料及临床表现
117例SMA患儿中,1型62例(53.0%),2型45例(38.5%),3型10例(8.5%),男女比=1.4∶1,门诊确诊占69.2%(81/117),住院确诊占30.8%(36/117)。患儿来自重庆41.9%(49/117)、四川35.9%(42/117)、贵州12.0%(14/117)、云南3.4%(4/117)及其他地区6.8%(8/117)。1型SMA起病、就诊、确诊年龄均早于2、3型SMA(P<0.05)。1型SMA就诊时间窗(起病年龄至就诊年龄)短于2、3型SMA(P<0.05)。各型性别构成比、确诊时间窗(就诊年龄至确诊年龄)差异无统计学意义(P>0.05)。
85.5%(100/117)的SMA患儿因肌无力首次就诊,14.5%(17/117)因肺炎首次就诊,后者多见于1型SMA(P<0.05)。所有SMA患儿均有肌张力减低、腱反射消失、运动里程碑发育落后,其中1型SMA均不能独坐,2型不能独站,3型可独走、随年龄增长行走能力逐步下降。SMA患儿还可出现抬头无力(47.9%,56/117)、脊柱侧弯(35.7%,41/115)、下肢关节挛缩(29.1%,34/117)、进食费力(21.4%,25/117)、哭声无力(18.8%,22/117)等。抬头无力、哭声无力、进食费力多见于1型SMA(P<0.05),2型SMA脊柱侧弯和下肢关节挛缩发生率高于1型(P<0.05),见表1。

表1 117例SMA临床资料的比较
2.2 SMN1基因检测结果
117例SMA患儿均为SMN1基因纯合缺失。7号外显子纯合缺失者68.4%(80/117),7、8号外显子纯合缺失者30.8%(36/117),8号外显子纯合缺失者0.8%(1/117)。SMN1基因缺失类型在各型SMA中的检出率差异无统计学意义(χ2=1.742,P=1.000)。
2.3 预后
中位随访时间为20.5个月(范围:3.0~50.7个月),随访期间死亡53例(45.3%),其中1型SMA 52例(98%,52/53),中位死亡年龄为6月龄(范围:1月龄至6岁);2型SMA死亡1例(2%,1/53),死亡年龄6岁;3型SMA无死亡病例。死亡原因均为重症肺炎并发呼吸衰竭。随访过程中所有患儿均有运动能力倒退,7例2型SMA有一过性运动功能进步,中位发生年龄为41月龄(范围:2岁3月龄至4岁7月龄)。至随访终点,所有2型SMA患儿均未能独走,1例已不能独坐;3型SMA患儿中1例已失去行走能力,9例有行走能力下降。
各型SMA 6年生存率差异有统计学意义(χ2=90.592,P<0.001),两两比较发现1型SMA 6年生存率低于2型SMA(χ2=75.315,P<0.001)和3型SMA(χ2=19.773,P<0.001),2型和3型SMA 6年生存率差异无统计学意义(χ2=0.304,P=0.581),见图1。1型SMA患儿在生后6个月、1年、2年、3年、4年、5年、6年生存率分别为53%±6%、28%±6%、21%±5%、17%±5%、15%±5%、15%±5%、10%±5%,而2、3型SMA在生后6个月至5年各时间点均无死亡,生后6年生存率分别为96%±4%、100%±0%。
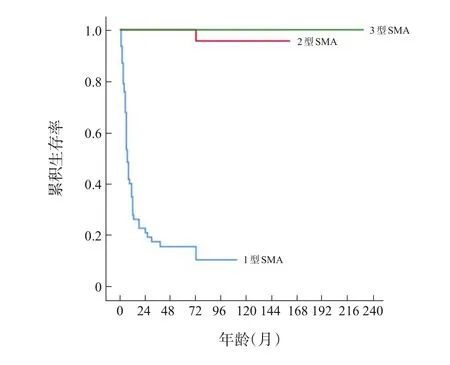
图1 不同类型SMA患儿的Kaplan-Meier生存分析图
2.4 1型SMA 6年生存率的危险因素
对62例1型SMA生存状况进行Kaplan-Meier单因素分析,结果显示,起病年龄≤3月龄、肺炎为首发症状、抬头无力可影响1型SMA 6年生存率(P<0.05),见表2。

表2 62例1型SMA Kaplan-Meier单因素分析
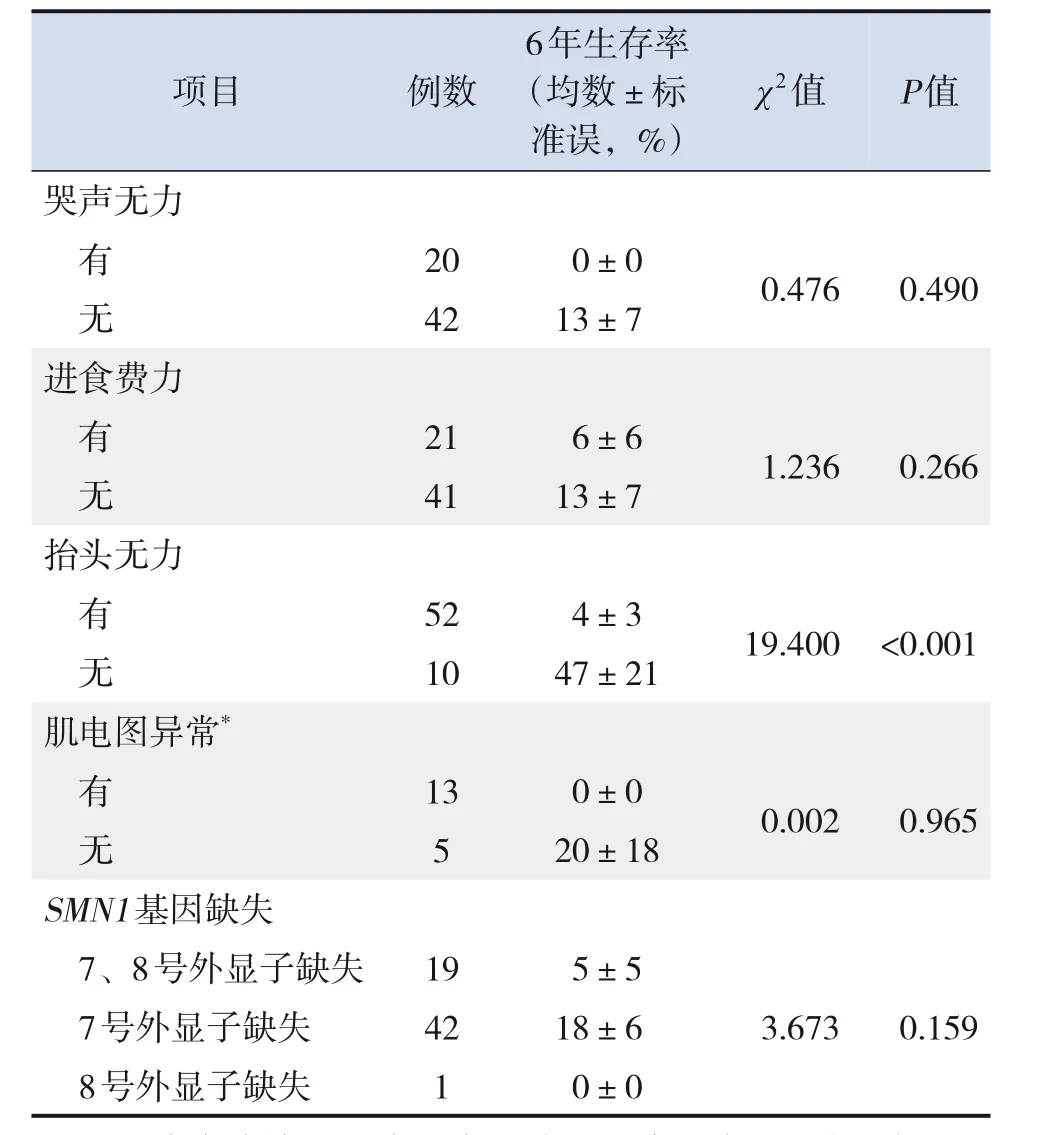
表2 (续)
进一步行多因素Cox回归分析,发现起病年龄≤3月龄、肺炎为首发症状、抬头无力是影响1型SMA 6年生存率的危险因素(分别OR=1.976、2.594、8.522,P<0.05),其死亡风险更高,见表3。
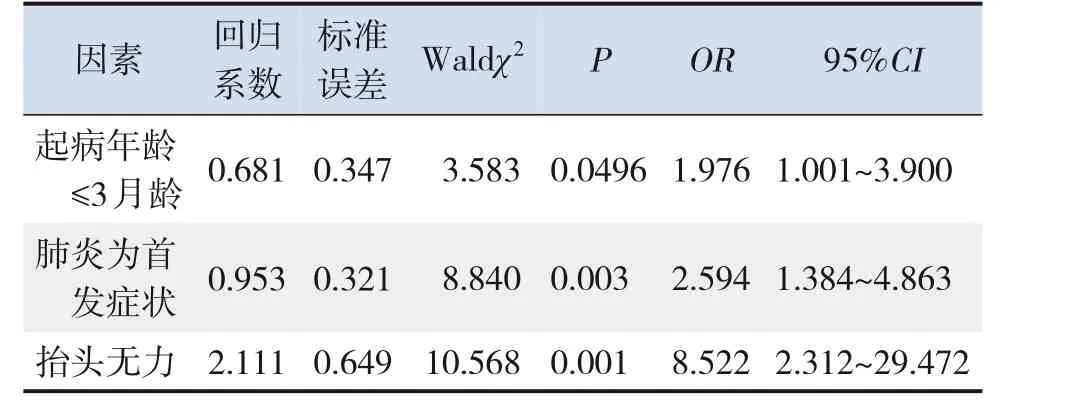
表3 1型SMA患儿生存时间的多因素Cox回归分析
3 讨论
SMA是婴幼儿时期最常见的致死性神经遗传性疾病。SMN1基因的缺失使SMN蛋白合成减少、脊髓前角运动神经元受损,导致患儿的运动功能进行性下降,最终丧失运动功能。临床上根据起病年龄及能够实现的最大运动功能将其分为4型,儿童主要为1~3型[3]。
本研究中1型SMA就诊时间窗明显短于2、3型,与Cao等[4]报道一致,考虑为1型SMA起病更早、更重,更容易被早期识别及诊治,2、3型SMA临床表现不典型、进展相对缓慢,容易被延误诊断。文献报道SMA确诊多在三级甲等医院[4],本研究中所有SMA均在我院确诊,确诊前曾辗转于各基层医院,因此提高基层医生对SMA识别能力,对减少SMA误诊、漏诊非常重要。
本研究中少数1型SMA患儿因肺炎首次就诊,肺炎易反复且迁延不愈,最终确诊为SMA。因此,当患儿反复发生迁延性肺炎,且四肢松软、运动里程碑落后时,临床应警惕SMA的可能。2型SMA较1型SMA更容易出现脊柱侧弯、关节挛缩,宜早期进行骨科评估及干预[5]。文献报道SMA还可累及心血管、消化、代谢、营养、发育等多个系统[6-7]。因此,对于SMA患儿临床上应予以全面评估和系统管理。
SMN1基因检测是诊断SMA的金标准[8]。本研究通过实时荧光探针定量PCR法行SMN1基因检测,SMN1基因纯合缺失率100%,7号(伴或不伴8号)外显子纯合缺失占99.1%,与王旻晋等[9]报道一致。既往报道中SMN1基因多以7号和8号外显子纯合缺失为主[10],本研究中7号外显子纯合缺失最为常见,可能与地域差异及实验室检测技术有关。本研究中仅有1例8号外显子纯合缺失,该患儿为2月龄男性,以肌无力起病、进行性运动功能倒退,5月龄死亡,临床经过符合1型SMA表现,李鸿等[11]有类似报道,但8号外显子的致病性及作用机制尚不清楚。SMN2基因与SMN1基因同源,SMN2基因的拷贝数可能与SMA的严重程度呈负相关[12-14],由于本研究为回顾性研究,限于当时经济条件及实验室技术,未能进行SMN2基因拷贝数检测。
进一步分析发现,1型SMA患儿生后1年、2年、5年生存率仅为28%±6%、21%±5%、15%±5%,低于德国、美国、我国北方及香港地区的1年、2年、5年生存率(36.9%~50.0%、30.8%~40.0%、24.6%~29.3%)[15-18],后者较高的生存率可能与家庭无创呼吸机的使用、肠内营养的支持有关[15]。本研究中所有SMA患儿均未接受基因修饰治疗,地处经济欠发达的西南地区,各型SMA患儿大多未能坚持规律随访及康复,且均无家庭呼吸机的使用,因此本研究中SMA的病死率更接近SMA的自然病死率。
本研究中1型SMA中位死亡年龄为7月龄,较国内外报道的8.0~13.5月龄[12,17,19]稍早。重症肺炎、呼吸衰竭是主要的死亡原因。因此,加强呼吸道管理是SMA患儿管理的重要举措,咳嗽辅助、肺部理疗及无创呼吸通气的使用可以有效提高SMA患儿的生活质量[20]。
本研究中少数2型SMA患儿有一过性的运动功能进步,其中位发生年龄为41月龄,至随访终点,所有SMA患儿均出现运动能力的倒退。Mercuri等[21]发现部分SMA患儿早期(<5岁)可出现一定程度的进步。Chabanon等[22]发现2、3型SMA患儿开始退步年龄在2岁、9岁。SMA的运动功能可呈非线性倒退,临床诊断时应警惕,以免误诊。
综上,SMA是一种严重的神经遗传性疾病,危害着婴幼儿生命及生存质量,对SMA早期识别、早期诊断、早期多学科综合管理[23-25],可一定程度上延长患儿的生存时间,改善患儿的生存质量。近年来基因治疗领域取得了重大突破,目前已有基因修饰药物Nusinersen、Risdiplam,基因治疗药物Zolgensma被美国食品药品监督管理局批准上市[26]。研究表明以上药物均可在一定程度上提高SMA患儿运动能力[27-30],但以上药物问世时间尚短,在真实世界的疗效与安全性有待进一步评估。未来,我们期待更多的SMA患儿能受益于基因治疗。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国生殖健康(2020年4期)2021-01-18 02:58:10
祝您健康·文摘版(2020年7期)2020-07-13 09:35:19
科学生活(2019年7期)2020-01-01 08:28:02
中国生殖健康(2019年2期)2019-08-23 08:11:52
养殖与饲料(2019年10期)2019-02-25 14:52:37
中国生殖健康(2018年4期)2018-11-06 07:12:16
山东畜牧兽医(2018年3期)2018-04-26 09:10:34
老年教育(老年大学)(2018年12期)2018-01-28 14:15:50
癌症进展(2016年12期)2016-03-20 13:16:14
