
HSD17B4基因突变致D-双功能蛋白缺乏症
2021-10-30 02:35:06杨树梅曹传顶丁颖王铭杰岳少杰
中国当代儿科杂志 2021年10期
杨树梅 曹传顶 丁颖 王铭杰 岳少杰
(中南大学湘雅医院新生儿科,湖南长沙 410008)
D-双功能蛋白缺乏症(D-bifunctional protein deficiency,DBPD)是由HSD17B4基因突变引起的D-双功能蛋白(D-bifunctional protein,DBP)活性低下或表达下降导致过氧化物酶体脂肪酸β氧化障碍而引起的一系列临床症状[1]。该病首次报道于1989年[2],发病率约1/10万[1]。迄今国外共报道163例,国内仅3例[3-4],目前国内对该病认识不足。本文报道1例新生儿期起病,HSD17B4基因突变所致DBPD的诊治过程,以利于提高临床医生对该病的认识,指导早期诊断。
1 病例介绍
(1)病史:患儿,男,15 d,因反复抽搐14 d就诊。患儿系试管婴儿,第3胎第1产,胎龄39+4周,剖宫产娩出,出生体重3 400 g,Apgar评分1、5、10 min均为10分。患儿生后即出现少哭、吸吮差、精神反应欠佳、肌张力低下。生后38 h收入当地医院新生儿科,体格检查时发现有频繁抽搐,表现为频繁眨眼、四肢强直抽动,每次持续1 min左右(具体抽搐次数不详),予以苯巴比妥15 mg静脉推注后无明显改善,入院10 h后(生后48 h)转入某儿童医院住院治疗。入住儿童医院后临床可见反复抽搐发作,表现为频繁眨眼,双眼凝视,四肢强直抽动,脸部涨红,口唇咀嚼样动作,伴心率、呼吸增快,偶有血氧饱和度下降至75%左右,持续约1 min自行缓解,每日发作10余次。查血常规、肝肾功能、心肌酶、血糖、血脂、电解质、血氨、血乳酸、C-反应蛋白、免疫全套、TORCH全套检查,结果均正常;脑脊液常规和生化正常,血培养及脑脊液培养均阴性;血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析未见异常。住院期间行颅脑MRI示双侧颞部外间隙稍宽,右侧额叶稍粗血管影,顶枕部头颅血肿;耳声发射双侧未通过,脑干听觉诱发电位示双侧听觉传导通路受累(中枢段为主)。入院后行24 h视频脑电图,监测到15次局灶运动性发作。给予苯巴比妥钠及左乙拉西坦抗癫痫治疗,效果不佳仍反复抽搐,表现同前。入院后(生后6 d)采集患儿及其父母静脉血外送(武汉康圣达医学检验所)行全外显子组测序,结果暂未回报。为求进一步诊治来我院。患儿母亲2010年因社会原因流产1次,2019年行试管婴儿因胎停流产1次。否认家族近亲婚配史,否认家族癫痫史。
(2)体格检查:体温36.5℃,脉搏161次/min,呼吸46次/min,血压68/47 mm Hg(平均动脉压54 mm Hg),经皮血氧饱和度94%,头围34 cm,体重3 130 g,身长50 cm。发育正常,无颅面部畸形,弹足底5下,无哭声、有皱眉动作,双侧瞳孔等大等圆,对光反射灵敏,体检时发现双眼频繁眨动,口唇咀嚼样动作,无鼻翼扇动。无三凹征,双肺呼吸音粗,听诊无干湿啰音。心率161次/min,心律整齐,心音正常,未闻及心脏杂音。腹部平软,肝肋下2 cm,质软,脾肋下未触及。脊柱正常,双上肢围巾征阳性(上肢环绕颈部,肘至腋前线),双腘窝角180°,肛门外生殖器无畸形。吸吮反射、觅食反射、拥抱反射、握持反射均未引出。
(3)辅助检查:入院后查血气分析、血常规、肝肾功能、心肌酶、血糖、电解质、C-反应蛋白、免疫全套、脑脊液常规和生化正常,血培养及脑脊液培养均阴性,头颅B超正常。入院当天行12 h视频脑电监测,监测到21次抽搐发作,发作期图形表现为:起始于Cz导联的低波幅快波节律,泛化至全脑弥漫性快波夹杂尖波、棘波发放,随后波幅渐低,频率渐减,为局灶继发全面性发作(图1)。发作间期可见多灶性癫痫波发放及间断爆发-抑制图形(图2)。同期临床表现为右下肢抽动,累及双下肢,随后全身肌肉持续强烈收缩,伴不对称偏转症状,头眼强制性向右偏转,右侧嘴角歪斜,面部表情不对称,伴咀嚼样动作、心率和呼吸增快,持续1~2 min。生后2个月11 d查血清极长链脂肪酸(very-long-chain fatty acids,VLCFA)显示二十六烷酸显著增高(2.91 nmol/L,参考值≤1.3 nmol/L),二十六烷酸/二十二烷酸比值轻微增高(0.06,参考值≤0.023)。
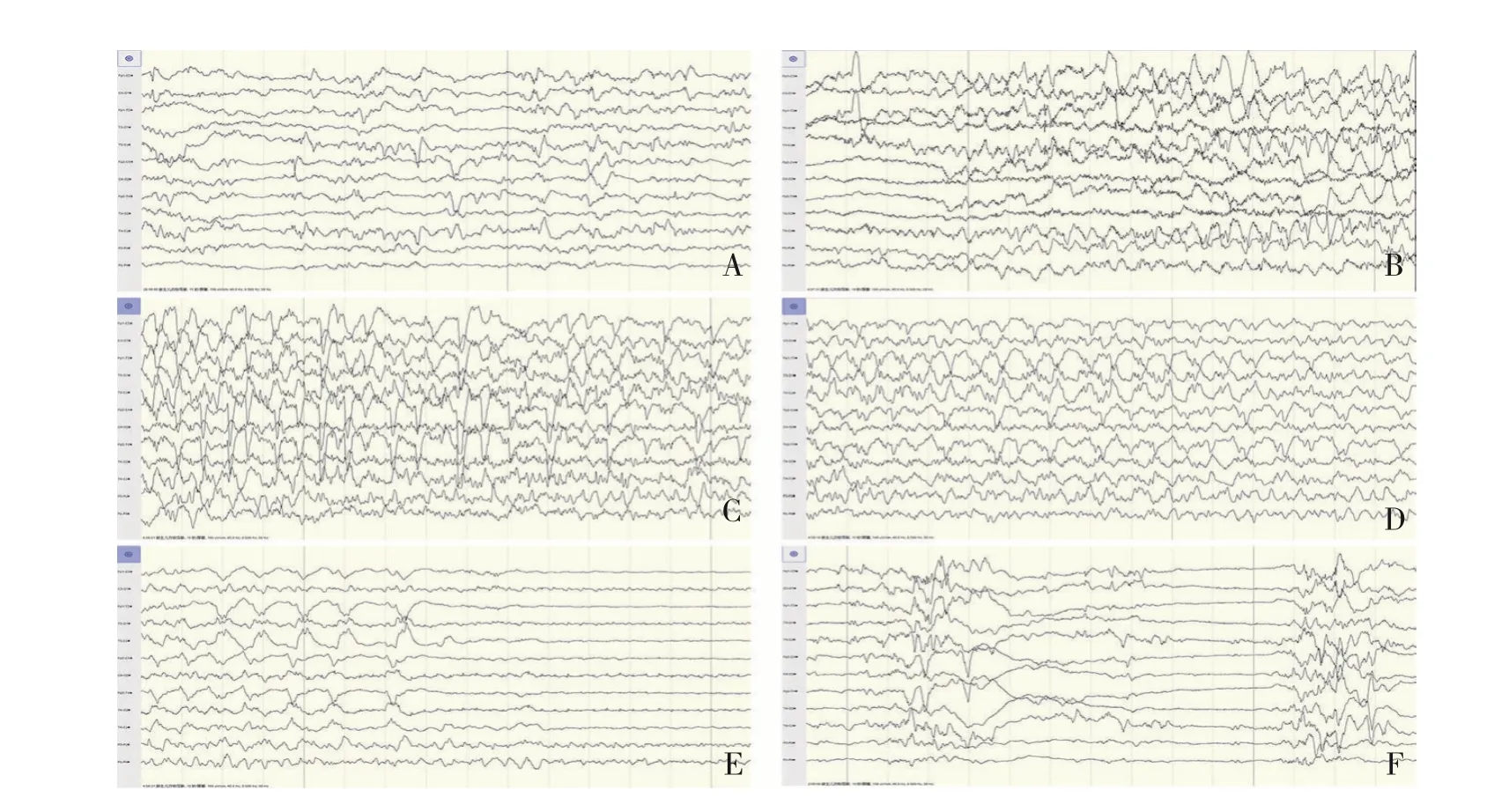
图1 患儿局部继发全面发作脑电图像
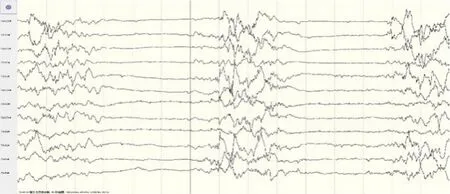
图2 患儿生后12 h视频脑电监测
(4)基因检测:生后6 d,患儿及其父母外周静脉血全外显子组测序(武汉康圣达医学检验所)显示,患儿HSD17B4基因存在c.111C>T(p.S37S)、c.1520_1532del(p.V507fs)复合杂合突变,其中c.111C>T(p.S37S)为同义突变,随后复测(深圳华大检验实验室)结果显示HSD17B4基因存在c.1448_1460del(p.Ala483Aspfs*37)、c.101C>T(p.Ala34Val)复合杂合突变(图3)。生后1个月26 d再次查患儿及其父母外周血CNVseq+染色体+线粒体及全外显子组测序(嘉检医学),结果显示,CNVseq+染色体+线粒体未见异常,全外显子组测序结果与华大检验实验室一致,故采用华大实验室结果。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)的变异解读指南[5],c.1448_1460del判定为致病性变异(PVS1+PM2+PM3):该突变导致氨基酸的移码突变,属于功能变异,符合PVS1;该变异在人类基因突变数据库(HGMD,http://www.hgmd.cf.ac.uk)及ClinVar数据库(https://www.ncbi.nlm.nih.gov/clinvar)中无收录,为ESP数据库(http://evs.gs.washington.edu/EVS)、千人数据库 (http://browser.1000genomes.org)及ExAC数据库(http://exac.broadinstitute.org)中正常对照人群未发现的变异,符合PM2;在其反式位置上检测到致病变异,符合PM3;该变异符合3条致病性证据。c.101C>T判定为疑似致病性变异(PM2+PM3+PP1+PP3+PP4):该变异为ESP、千人数据库及ExAC数据库中正常对照人群未发现的变异,符合PM2;在反式位置上检测到致病变异,符合PM3;突变与疾病在家系中共分离,符合PP1;多种生物信息学方法预测出该变异会对基因或基因产物造成有害的影响,符合PP3;变异携带者表型高度符合HSD17B4单基因遗传病,符合PP4;该变异符合5条致病性证据。
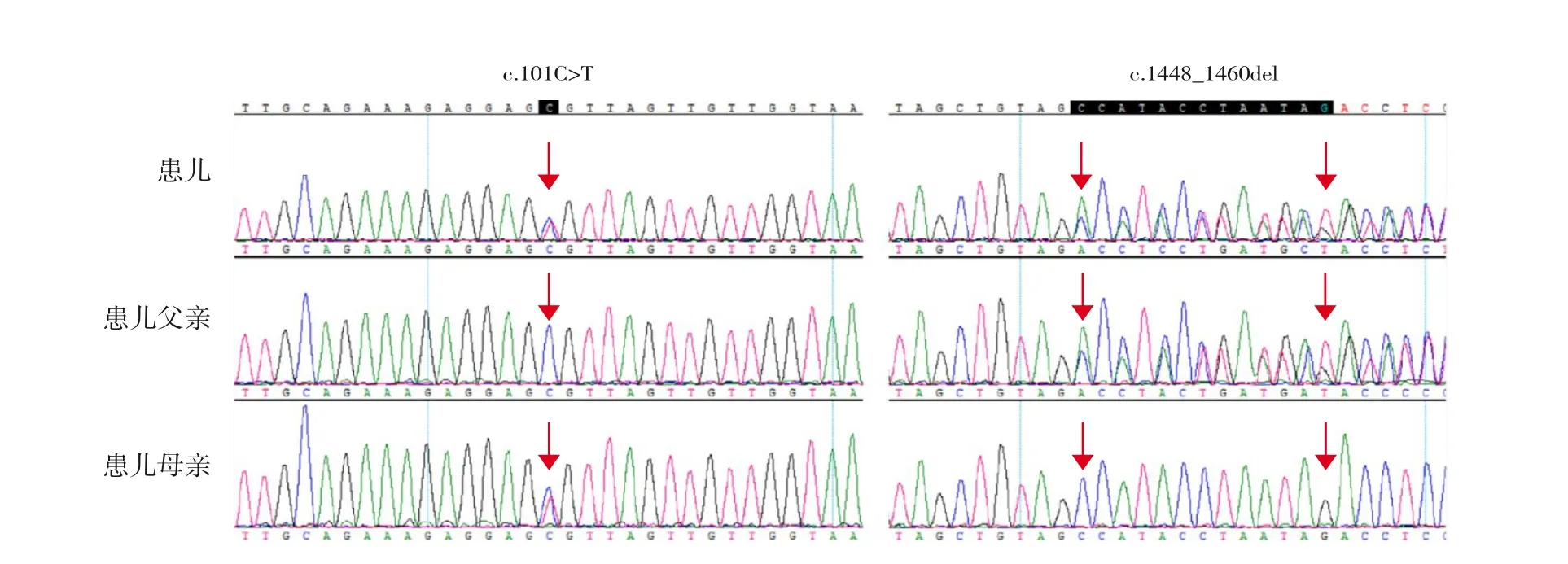
图3 患儿及其父母HSD17B4基因Sanger测序结果
2 临床经过
患儿入院后增大左乙拉西坦剂量[加量至38 mg/(kg·d),2次/d],同时维生素B6诊断性治疗及对症支持治疗,经治疗后抽搐仍无明显改善。入院当天视频脑电图监测到患儿频繁癫痫发作(21次),继续增加左乙拉西坦至57.5 mg/(kg·d),2次/d。5 d后复查12 h脑电图仍有频繁发作(13次),当天加用咪达唑仑[2μg/(kg·min)]持续静脉滴注及奥卡西平[起始剂量9.3 mg/(kg·d),2次/d],逐渐增加至16.9 mg/(kg·d),2次/d口服。调整药物9 d后,12 h脑电图结果较前好转,逐渐减停咪达唑仑。继续采用左乙拉西坦联合奥卡西平治疗,因脑电图无改善,逐渐增加奥卡西平剂量,至24.1 mg/(kg·d),2次/d,后抽搐发作较入院时明显减少,遂出院。住院28 d出院,此时基因结果未回报,出院诊断:(1)难治性癫痫,(2)大田原综合征(Ohtahara syndrome,OS)可疑(追踪)。出院后继续服用左乙拉西坦[48.3 mg/(kg·d),2次/d]及奥卡西平[24.1 mg/(kg·d),2次/d]。
出院半月后(生后1个月19 d)患儿仍有频繁抽搐发作,入住我院小儿神经专科住院治疗,复查脑电图仍可见多次局灶性发作,加用托吡酯[1.6 mg/(kg·d),2次/d]并 减 量 左 乙 拉 西 坦[30 mg/(kg·d),2次/d],联合奥卡西平[30 mg/(kg·d),2次/d]抗癫痫治疗。1周后患儿出现进行性血小板下降,考虑与左乙拉西坦不良反应有关,于是将左乙拉西坦减量至15 mg/(kg·d),2次/d,同期脑电图较前明显好转,奥卡西平减量至24 mg/(kg·d),2次/d,托吡酯用法同前。调整药物后1 d患儿再次出现多次发作,遂将奥卡西平加量至30 mg/(kg·d),2次/d,托吡酯加量至2.8 mg/(kg·d),2次/d,左乙拉西坦逐渐减停。1周后血小板恢复正常值,未见明显临床发作,病情好转出院。住院23 d出院,此时复查基因测序及血清VLCFA结果未回报,出院诊断:OS。出院带药奥卡西平[30 mg/(kg·d),2次/d]及托吡酯[2.8 mg/(kg·d),2次/d],门诊随访调整药物剂量。
患儿11月龄时回门诊随访,体检:身长73 cm(P25),头围45 cm(P50~P75),体重7.59 kg(P3)。肌张力低下(双上肢环绕颈部,双足跟至耳、双腘窝角180°),无追物、追声,不能竖头、抬头,不能独坐。贝利婴幼儿量表检测:智力发育指数为1分(家属诉偶可逗笑),运动发育指数为0分,提示存在严重智力及运动发育迟滞。复查3 h视频脑电图未见临床发作及电发作,但仍有多量多灶性癫痫波发放。半月后(11.5月龄)电话随访得知患儿死亡(具体过程不详)。
3 诊断思维
患儿病例特点:(1)足月男孩,出生无窒息史,生后即起病,主要表现为难治性抽搐发作,伴有持续反应吸吮差、肌张力低下;(2)双侧脑干诱发电位减弱,脑电图监测到频繁局灶运动性发作及爆发-抑制图形;(3)血生化、感染指标、脑脊液检查、头颅MRI、血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析等未见异常。
根据脑电图结果及相关临床表现,临床初诊为新生儿惊厥。引起新生儿惊厥的常见病因有缺氧缺血性脑病、颅内感染、颅内出血、脑梗死、先天性脑发育畸形、电解质紊乱、低血糖;此外,胆红素脑病、低血糖脑病、新生儿戒断综合征、遗传代谢性疾病、良性家族性新生儿惊厥、OS等也可引起新生儿惊厥。本例患儿出生时无窒息史,脑脊液常规、生化、培养检查及头颅MRI均未发现明显异常,可排除缺血缺氧性脑病、颅内感染、颅内出血、脑梗死和先天性脑发育畸形等病因。患儿无严重高胆红素血症和低血糖病史,多次查血糖、电解质、胆红素均正常,可排除电解质紊乱、胆红素脑病、低血糖脑病等原因。母亲孕期无特殊用药史,可排除新生儿戒断综合征。患儿生后即出现反应吸吮差、肌张力低下,脑电图有爆发-抑制图形,不支持良性家族性新生儿惊厥等。血糖、血氨、血乳酸、血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析基本正常,不支持常见遗传代谢性疾病,但有少数遗传代谢病可见正常血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析结果,比如吡哆醇依赖性癫痫[6]、Zellweger综合征[7]、新生儿肾上腺脑白质营养不良[8]等有待进一步排除。OS的主要临床及电生理特征为难以控制的强直-痉挛发作和爆发-抑制脑电图,脑电图是其重要确诊依据;OS可能病因为脑结构异常、代谢紊乱和基因突变。本病患儿主要临床表现为生后难以控制的频繁抽搐发作,发作形式为局灶运动性发作,脑电图睡眠期及清醒期均可见爆发-抑制图形,符合OS典型临床及电生理表现,临床主要考虑OS。但Zellweger综合征、新生儿肾上腺脑白质营养不良等过氧化物酶体缺陷代谢遗传病,临床也可表现为频繁抽搐发作及肌张力减退,常规血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析通常无异常,但其生化特征性表现为血清VLCFA增高,其致病基因与OS不同。该患儿生后2 d头颅MRI未见明显异常,常规血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析无异常,因此,外送血清VLCFA及基因检测明确诊断。出院后血清VLCFA结果回报显示二十六烷酸显著增高,基因结果回报提示HSD17B4基因存在致病性c.1448_1460del、c.101C>T复合杂合突变,提示DBPD。回顾本例患儿诊治过程发现,DBPD与OS的临床表现与神经电生理表现存在高度相似性,传统临床诊断方法难以辨别,极易出现误诊。血清VLCFA增高及HSD17B4基因突变为DBPD特异性改变,对临床高度怀疑“OS”的患儿,应常规进行血清VLCFA及基因检测,及时明确诊断。
4 诊断及诊断依据
诊断:DBPD。诊断依据:(1)生后即有反应差、吸吮差、肌张力减退、难治性癫痫发作,符合该病相关临床表现。(2)耳声发射及脑干听觉诱发电位检查均提示双侧听力受损,符合该病神经电生理表现。(3)血清VLCFA检测结果异常增高,符合该病生化表现。(4)患儿5号染色体HSD17B4基因存在c.101C>T和c.1448_1460del突变,经家系验证为复合杂合突变,符合该病遗传学特征。其中诊断依据(3)与(4)最具有诊断价值。
5 讨论
DBPD是一种罕见的过氧化物酶体单个酶缺陷遗传病,因HSD17B4基因突变所致DBP活性低下及表达下降引起的一系列临床症状[1]。DBP包含的功能单位2-烯酰辅酶a水合酶及3-羟基酰基辅酶a脱氢酶参与β氧化过程中的第二步水化反应和第三步脱氢反应[9-10],β氧化障碍可致毒性VLCFA堆积至全身各个器官引起一系列临床表现。DBPD分为3种类型:DBP完全缺乏型(Ⅰ型)、D-3-羟脂酰辅酶A水化酶缺陷型(Ⅱ型)、D-3-羟脂酰辅酶A脱氢酶缺陷型(Ⅲ型)[1,10]。不同类型预后不一,Ⅰ型患者常在1岁内死亡;Ⅱ、Ⅲ型患者生存期常超过10年。本例患儿不足1岁死亡,推测I型可能性大[1]。
该病起病年龄一般在新生儿期,进展迅速,临床表现表型多样,表现为出生时常存在颅面部发育畸形(巨颅、前额膨隆、囟门宽大、高腭弓、小下颌、马蹄足等)、肌张力明显减退、顽固癫痫发作、视听觉障碍、肝大及脑发育异常。患儿在我院两次出院诊断均为“OS”,需重点关注DBPD与OS的鉴别诊断。
该病相关文献多为个案报道及小样本研究,目前尚无统一规范的诊断标准,总结归纳已发表病例及回顾本文病例:(1)主要临床表现:肌张力减退(98%,83/85),生后1个月内出现癫痫发作(93%,78/84),生后1个月内出现肌张力减退合并癫痫发作(93%,79/85),颅面部畸形(67%,53/79),肝大(44%,32/73),听觉障碍(45%,29/64)[1]。OS一般在婴儿早期或新生儿期起病,主要临床特征为难以控制的癫痫发作,发作类型多样,多表现为强直-痉挛发作,也可为局灶运动性发作。本例患儿新生儿期起病,存在频繁及难以控制的癫痫发作(局灶运动性发作为主),这些表现同时是OS的典型临床表现,临床上极易误诊。但该患儿同时存在严重肌张力减退及双侧听力受损,根据已报道病例得知,肌张力严重减退、颅面部畸形、肝大与视听觉障碍等均为DBPD特征性表现,OS并无相关报道,可鉴别。(2)常见神经影像学改变:皮质发育不良(27%,12/45)、脑室系统扩张(30%,16/53)、白质成熟延迟(35%,17/48)、大脑半球脱髓鞘(19%,9/47)、小脑半球脱髓鞘(6%,3/47)、小脑萎缩(16%,8/49)[1,11]。本例患儿生后2 d头颅MRI未见明显异常,有文献报道生后7 d头颅MRI无异常,但生后31 d复查可见双侧大脑半球及胼胝体发育不良的DBPD患儿[3]。提示该病头颅结构性异常可能为晚发进展性,必要时需复查。OS神经影响学检查常有明显异常,如脑发育不良(巨脑回、多小脑回、脑白质或胼胝体发育不良等)、脑萎缩、先天性脑积水等[12],同时KCNQ2、SCN2A、STXBP1等基因突变所致OS可不伴头颅MRI异常[13-14]。二者影像学均无特征性改变,无法相鉴别。(3)临床电生理表现:双侧脑干诱发电位减弱(90%,19/21),视网膜电图活动减弱(77%,17/22),神经传导速度延迟(67%,16/24)[1]。OS主要电生理特征为典型爆发-抑制脑电图,脑电图是OS的重要诊断依据。本例患儿脑电图表现为睡眠期及清醒期均存在爆发-抑制图形,根据脑电图表现极易误诊为OS。但该患儿同时存在双侧脑干诱发电位减弱,回顾已报道文献得知,90%DBPD患者神经电生理表现为双侧脑干诱发电位减弱,仅1例报道脑电图为爆发-抑制图形[15],而OS无脑干诱发电位异常。(4)生化特征:血清VLCFA是DBPD特异性生化指标[16],可与OS鉴别。本例患儿生后2 d血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析无异常,2月龄查血清VLCFA异常增高。KCNQ2、SCN2A、STXBP1等基因突变所致OS可不伴常规血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析异常,常规血氨基酸和酰基肉碱串联质谱分析、尿有机酸分析无法鉴别DBPD与OS,但VLCFA是DBPD特征性改变,OS无血清VLCFA异常。临床怀疑该病时,应尽早行血清VLCFA检测辅助诊断及鉴别。(5)基因检测:HSD17B4基因变异是该病主要致病原因。目前已知与OS相关的基因包括ARX、SLC25A22、KCNQ2、SCN2A、KCNT1及STXBP1等[17],查阅相关遗传数据库,HSD17B4基因不属于OS基因谱。本例患儿全外显子测序结果提示HSD17B4基因存在c.1448_1460del、c.101C>T复合杂合突变,其中c.1448_1460del为首次报道的新发突变,基因检测是唯一确诊手段。
该病治疗上尚无显著进展,以对症支持治疗为主,早期诊断可进行相关风险评估,并指导遗传咨询和产前诊断。HSD17B4基因突变所致新生儿起病DBPD临床及脑电表现与OS相似,临床极易误诊,怀疑该病时应详细评估肌张力、颅面部发育、肝脏及视听觉情况等临床特征,重视脑干诱发电位、视网膜电图、脑电图及肌电图等神经电生理及头颅MRI检查,及时进行特异性生化指标及遗传学检测明确诊断。血清VLCFA是该病支持诊断的特异性生化指标,基因检测是唯一确诊手段。
猜你喜欢
中国现代神经疾病杂志(2017年1期)2017-03-29 06:39:34
家庭百事通·健康一点通(2017年3期)2017-03-22 20:13:17
现代电生理学杂志(2016年1期)2016-07-10 10:20:58
广东药科大学学报(2016年6期)2016-03-10 07:33:32
动物营养学报(2015年9期)2016-01-07 11:29:44
川北医学院学报(2015年5期)2015-12-05 08:22:33
现代电生理学杂志(2015年1期)2015-07-18 11:02:17
中国当代医药(2015年7期)2015-03-01 02:01:13
放射学实践(2015年2期)2015-02-14 05:38:58
高中生学习·高一版(2014年6期)2014-07-05 10:20:16
