
基于深度学习的药物设计方法
2021-10-27 08:25:10李风雷胡乔宇熊若凡白芳
自然杂志 2021年5期
李风雷,胡乔宇,熊若凡,白芳
上海科技大学 免疫化学研究所,上海 201210
随着科学技术的不断进步,药物研发已经从“偶然发现”模式逐渐转型为“理性设计”模式。特别是计算机辅助药物设计(computer aided drug design,CADD)方法的出现,极大地丰富了“理性设计”技术,作为与实验技术互补的关键手段,不断推动着创新药物的研发进程。
药物研发过程可简化为4个主要阶段:靶标的鉴定、先导化合物的发现与优化、临床前研究以及临床研究。首先,基于特定的疾病类型,通过遗传学、组学以及生物信息学等方法,发现和鉴定与该疾病的发生、发展以及恶化过程直接相关的分子靶标,通过一系列评估确定药物靶标。其次,基于已知的药物靶标,结合结构生物学、计算机辅助药物设计、药物化学、分子生物化学、药理评价等方法,寻找先导化合物,再通过不断修饰化学结构,优化其在有机体内的吸收、分布、代谢、排泄和毒性等性质。再次,对于具有良好成药性和体外实验中有效的先导化合物,进一步开展临床前研究,在实验动物、人体类器官上验证先导化合物的药效和药代动力学性质,这一过程通常是由学术界和工业界合作完成的。最后,经过系统评估,只有在临床前研究中药效与安全性均佳的候选化合物,才能被推向临床研究与评估。临床研究通常也分为4个阶段:主要测试药物安全性的临床I期;在相对较小的样本上测试药物有效性的临床II期;在大样本上测试药物有效性的临床III期;以及上市后长期观察的临床IV期。
近年来,生物制药行业的蓬勃发展,产生并积累了越来越多的药物研发数据,这为人工智能技术在药物研发行业的渗入铺平道路。传统的CADD在药物研发的整个周期特别是临床前研究中具有重要的作用。20世纪90年代以后,大量药物设计计算方法不断被提出,助力着药物研发的各个阶段。生物医学大数据的涌现和人工智能技术的革新,无疑将进一步促进CADD方法的发展。本文将针对人工智能技术(主要是深度学习算法)在药物设计方法开发中的应用,进行简要回顾和总结,以期为药物设计提供更多的思路和方法。
1 人工智能与深度学习技术
人工智能(artificial intelligence,AI),是指人工制造的机器系统所表现出来的智能。该词语最早由麻省理工学院的约翰·麦卡锡于1956年在达特茅斯(Dartmouth)会议上提出。人工智能的发展跌宕起伏,并于近年开始加速。特别是大数据的出现、并行计算能力的提升和先进算法的提出,使得人工智能的发展进入前所未有的炙热局面。
机器学习是实现人工智能的一种方式,是人工智能的子领域。机器学习基于已有的数据、知识或者经验,自动识别和解析(“学习”)数据,总结有意义的模式,并以此在相似的环境里做出预测或决策。机器学习可分为:有训练标签的监督学习(supervised learning)、无训练标签的无监督学习(unsupervised learning)、通过观察环境做出动作并获得环境奖励的强化学习(reinforcement learning)、有部分训练标签的半监督学习(semi-supervised learning)、交互式获得标签的主动学习(active learning),以及学习如何学习的元学习(meta learning)等。
深度学习(deep learning)是机器学习的关键技术之一。近10年来,深度学习技术一路高歌猛进,在机器视觉、自然语言处理、机器翻译和路径规划等领域均取得了令人瞩目的成绩。人工智能作为引领未来的战略性技术,在生命科学特别是药物设计领域的应用得到逐步推广,对药物设计方法的研究起着重要的推动作用。本文将简要介绍一些重要的深度学习算法,以及这些算法在药物设计计算方法中的应用。
2 神经网络算法
神经网络算法是深度学习的一个重要的代表性算法,设计灵感来自生物神经网络。神经网络算法从信息处理角度对生物神经元网络进行抽象,建立神经元之间的通信网络,神经元间的不同连接方式组成不同的学习网络。神经元将输入的“信号”处理并逐层传递,最终获得输出。以下介绍几种简单的神经网络形式。
2.1 多层感知机
多层感知机(multi-layer perceptron,MLP)是由一个输入层(input layer)、一个或多个隐藏层(hidden layer)和一个输出层(output layer)构成的全连接网络(图1)。全连接网络中每个神经元都与前一层的神经元相连接,且连接均具有权重。因此,每个神经元可由前一层的所有神经元计算得出,公式如下:

图1 多层感知机
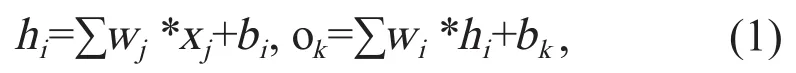
其中,xj是输入层的神经元,hi是隐藏层的神经元,ok是输出层的神经元。为了避免网络输入与输出间线性依赖的局限性,感知机的神经元中还可引入非线性激励函数(如Sigmoid、tanh和Softplus等),使得神经元的输入以非线性的形式映射到输出端。
2.2 卷积神经网络
卷积神经网络(convolution neural network,CNN)一般由卷积层和池化层组成。卷积计算作为CNN中最重要的部分,可有效地减少神经网络中的参数。如图2所示的网络由两层卷积层构成,每一层中的每一步操作均为一个卷积,用公式表示为:
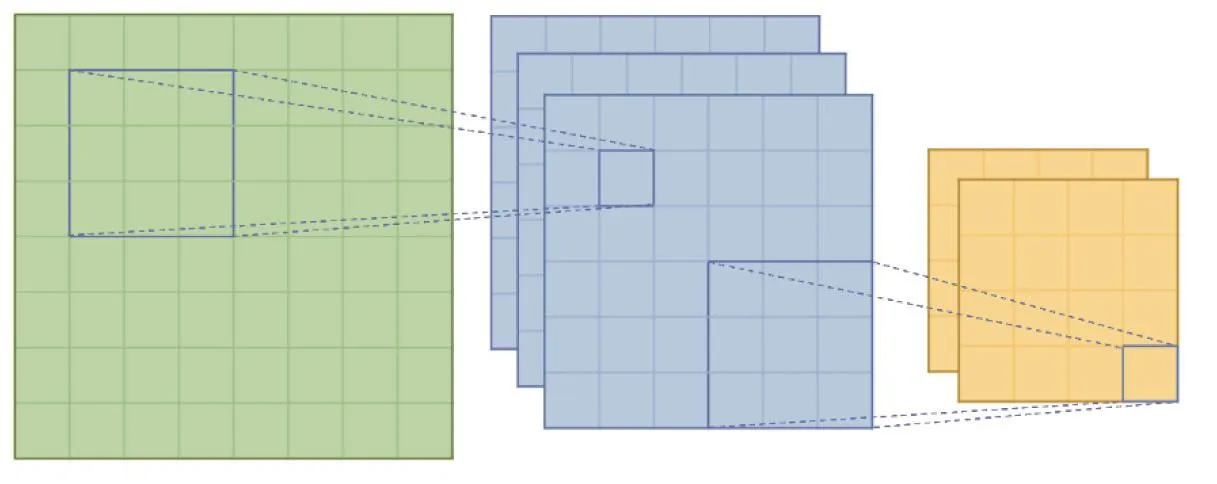
图2 卷积神经网络
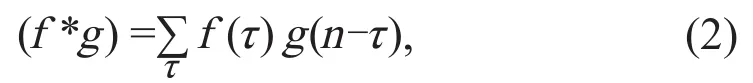
其中,f为输入数据,g为卷积核。该公式为数学定义的卷积,且是一维形式,而在深度学习中,往往用到的是高维的形式,并且在实现的时候会做一些修改。一层的卷积操作即是一个卷积层。增加卷积的层数、使用残差网络和池化等操作,可进一步优化卷积神经网络(如VGG、Resnet等)。
2.3 循环神经网络
循环神经网络(recurrent neural network,RNN)是考虑时间前馈的神经网络。RNN考虑了输入的序列性,即每一次输入都考虑到之前输出的信息,体现出“记忆功能”,是进行实践序列分析最好的选择(图3)。其模型表示为:
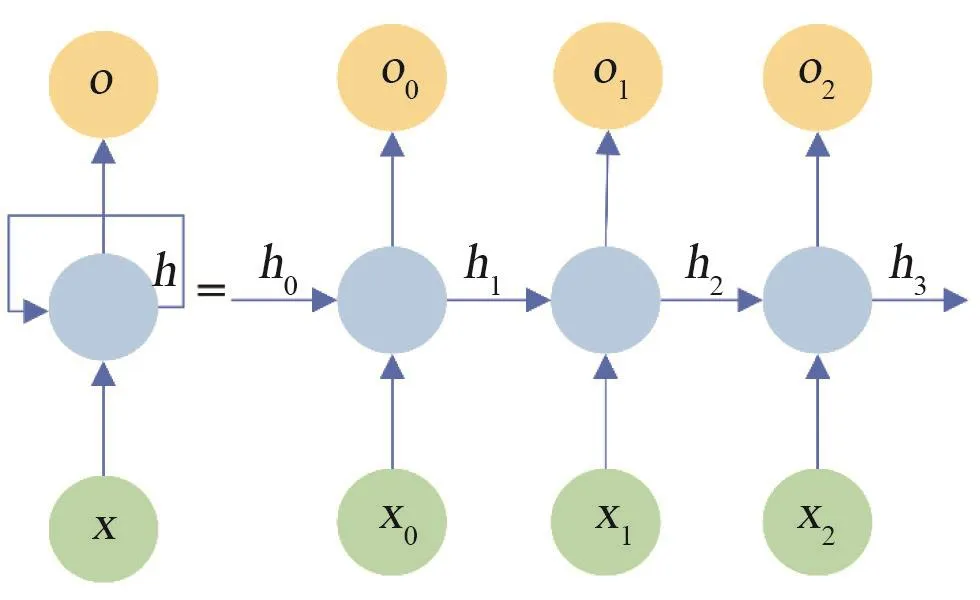
图3 循环神经网络
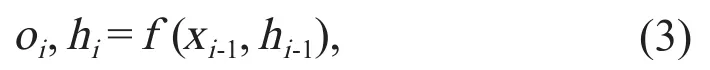
其中,xi是输入层的神经元,hi是隐藏层的神经元,oi是输出层的神经元。对当前学习之前的信息有选择性地输入,可提高RNN的效果,如长短期记忆(long short-term memory,LSTM)、门控循环单元(gated recurrent unit,GRU)等。
2.4 图神经网络
图是一种具有点和边的结构数据。图神经网络(graph neural network,GNN)则是一类基于深度学习的处理具有图信息的方法(图4),由于其较好的性能与可解释性,已经成为一种应用广泛的图分析方法。图神经网络的数学模型可表示为:

图4 图神经网络
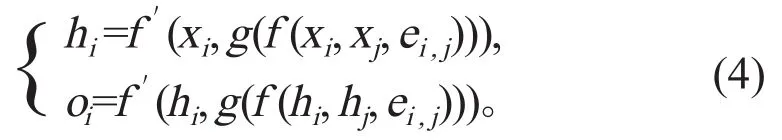
第i个点的特征hi是通过与其相邻的所有xj点的特征、xi本身的特征和其与xj间的边ei,j的性质综合得出的。f 和f'是可微函数,g通常为求和、求均值或求最大值函数等。通过将状态hi与特征ei,j传递给g函数,来计算GNN的输出oi。
2.5 注意力机制
注意力机制(attention mechanism)源自人类视觉和大脑处理信号的机制。在认知中,人类往往会因为信息庞大而选择性地关注其中高价值的一部分而忽略其他信息,即注意力机制。处理数据时,注意力机制模型对数据构成元素引入了查询(query)、键(key)和值 (value)构成数据对,通过计算查询与键之间的相关性或者相似性,得到键对应于值的权重系数(图5)。注意力机制可描述为:

图5 注意力机制
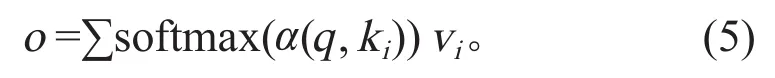
其中:q(query)在匹配ki(key)后与vi(value)结合,从而增加需要“注意力”部分的权重;α是注意力打分函数,不同的打分函数有不同的效果。在注意力机制模型基础上,衍生出Transformer[1]、Bert[2]等更高级的网络结构。
3 常用深度学习工具
目前已有多种深度学习框架,如PyTorch、TensorFlow、Paddle和Keras等,为神经网络的搭建提供了平台,给开发基于深度学习的应用模型带来极大便利。截至目前,已有多个基于深度学习算法的工具包,如DeepChem[3]、DeepPurpose[4]和OpenChem[5]等,在此基础上结合简单的深度学习算法的成功建立,为用户采用新的数据集进行训练提供了直接的工具。
4 深度学习算法在药物设计中的应用
药物研发过程主要包括药物靶标发现、先导化合物的发现与优化、候选药物的确定和成药性优化等步骤。近年来,深度学习在药物研发中的应用日渐广泛,以下将分别介绍深度学习算法在药物研发各步骤中的应用现状。
4.1 深度学习在药物靶标发现中的应用
药物靶标发现是现代药物研发模式中的第一步,也是决定新药研发成功与否的关键步骤。传统的药物靶标发现方法,主要是通过分析基因组学、蛋白质组学数据寻找疾病相关的潜在靶标。这些靶标可能是蛋白质、核酸(DNA、RNA)或其他生物大分子。随后,研究者使用细胞生物学、遗传学以及分子生物学的方法来验证潜在靶点的可成药性,包括靶标的功能机制、靶标与疾病的相关性和药物分子可设计性等,进而确定药物靶标。已经报道的药物靶标发现的计算方法主要分为两类:①基于反向分子对接的策略(如IdTarget[6]和TarFishDock[7]),该类方法计算量大,且受限于候选靶点结构的准确性,对于结构未知的体系无法准确预测;②以相似化合物具有相似的作用靶点为前提,通过比较发现的活性化合物与已知靶点的活性化合物的结构相似性,建立化合物与靶点之间间接关联网络,从而揭示候选药物靶点的方法(如ChemMapper[8]、PharmMapper[9]和SwissTargetPrediction[10]等),该类方法依赖于小分子-靶标信息的数据,因此在化学结构新颖的小分子上效果较差。除此之外,还有针对潜在靶点的成药性位点发现与评价方法(如Fd-DCA[11]等)也存在一定的局限性。基于深度学习的算法可以有效地综合来自多种数据集的信息,进而给出更加准确的预测,如DeepDTnet[12]根据构建的药物-基因-疾病网络就使用了图表示学习来进行靶标识别。通过采用该方法,研究者预测发现了Topotecan是ROR-γt(retinoic-acid-receptor-related orphan receptor-γt)的抑制剂,并得到实验的验证。这些方法的出现,无疑加速了药物靶点的发现与评价进程。
蛋白质-蛋白质相互作用是一类重要的药物靶点,已经有多种针对蛋白质-蛋白质相互作用的药物成功上市。然而,预测蛋白质-蛋白质相互作用仍富有挑战性。2019年,David Baker等[13]利用蛋白氨基酸序列作用位点的共进化理论与分子对接相结合的方法,通过发展计算方法成功预测了1 618组大肠杆菌蛋白对和384组未见报道的结核杆菌蛋白质-蛋白质相互作用对。然而,对于包括人类在内的真核生物而言,共进化分析所具有的同源序列信息较少。因此,基于统计算法的共进化分析方法的预测准确性便受到了极大的限制。深度学习方法对该领域的发展起到一定的推动作用(如DPPI[14]使用了卷积、随机投影和全连接预测3个模块构成的神经网络),通过对蛋白质-蛋白质作用对的序列氨基酸组成、顺序以及互作结构域序列的共同性特征等进行学习,发展了只依赖于氨基酸序列预测蛋白质-蛋白质互作的计算模型,在测试中其精度-回归曲线auPR得分约41%(人源测试集)。另外,如MaSIF[15]使用了几何神经网络(geometric neural network),将蛋白质表面的几何特征、化学特征与生物大分子间的互作关系相联系,建立了蛋白质-蛋白质互作以及蛋白质-小分子互作位点的预测方法,在蛋白质-蛋白质互作位点预测上每个蛋白的ROC AUC的中位数为0.81。
传统的蛋白质功能预测方式基于相似序列具有相似功能的进化理论,利用蛋白质之间的序列相似性,通过寻找与靶标蛋白序列相似,且功能明确的同源蛋白来间接推测其功能(如BLAST[16]、PSI-BLAST[17]、HMMER[18]等)。近年来,一些研究开始通过综合学习序列信息和已知的蛋白质结构,发展预测模型来预测蛋白质的功能。在2019—2020年蛋白功能预测大赛(critical assessment of functional annotation,CAFA)中,DeepGO[19]对蛋白序列编码后使用卷积神经网络获得潜在编码,并通过STRING[20]数据库中的蛋白-蛋白互作网络为每个蛋白生成图嵌入编码,随后将这两种编码合并后送入分类层,从而预测功能。TALE[21]使用基于自注意力(selfattention)Transformer来捕捉序列中的全局特征,进一步提高了对蛋白质功能预测准确性(其精度-回归曲线下面积AuPRC在不同的测试集和GO的不同子类中均比DeepGO高)。特别地,作者声称该方法对于同源序列信息缺乏的体系具有一定的优势。
4.2 深度学习在先导化合物的发现与优化中的应用
药物设计中,先导化合物的发现途径主要分为基于配体和基于靶标受体的两种药物设计方法。基于配体的药物设计,认为具有相同理化性质或结构的化合物应具有相同或相似的作用靶点及活性。因此,该方法策略是基于已知的活性化合物配体的结构及其活性信息,通过建立其结构与药效关系模型,来预测和评价新化学结构的相关生物学活性。其中,定量构效关系(quantitative structure-activity relationship,QSAR)是基于配体药物设计的主要方法之一,以配体(药物)的化学结构标识符和活性作为输入,通过多元线性回归、偏最小二乘回归和小波核偏最小二乘回归等方法,建立化学结构标识符与配体活性之间的关系。传统的QSAR方法依赖于大量的配体-活性关系数据,同时受限于已知活性配体的化学结构空间。因此,传统的QSAR方法具有较大的改进空间。近年来,深度学习的发展显著提高了QSAR方法的准确性和鲁棒性。Ma等[22]使用全连接的深度神经网络作为构建QSAR模型的方法。之后,他们又改进为多任务深度神经网络,通过训练不同任务的组合,他们发现,如果辅助任务的训练集与主任务的测试集的分子有较大的相似性,且两个任务的目标有生物活动关联性时可以提高主任务的预测,若没有关联则会降低预测,而若两个任务的数据集不同时对主任务的影响不大。
除基于配体的药物设计策略之外,基于结构的药物设计也是药物研发中的重要策略之一。该策略的传统的流程为:获取药物靶标的三维结构—确定药物靶标结构中的活性位点—使用分子对接等方法进行化合物的虚拟筛选。
随着结构生物学的发展,获得药物靶标三维空间结构的方法已经日渐成熟。如,AlphaFold[24]使用残差卷积神经网络,进行多序列比对(multiple sequence alignment,MSA)求出残基之间的距离矩阵,据此求出势能并通过Rosetta[25]获得结构。TrRosetta[26]在氨基酸距离矩阵的基础上,引入氨基酸构象角度矩阵,进一步提高结构的预测准确度。近日,AlphaFold2[27]的出现,实现了蛋白质结构领域的极大突破,并在CASP14(critical assessment of techniques for protein structure prediction 14)上的表现一骑绝尘,在一些较难体系中,其全局距离测试得分(global distance test score)为90,而其他队伍仅为75分左右!
RNA的结构预测一般集中在二级结构预测上,如MXfold2[28]使用卷积双向LSTM等网络层,输入RNA序列,输出相对碱基处于4种不同折叠状态的得分,然后使用动态规划预测最优二级结构。SPOT-RNA[29]与MXfold2的输入和网络相似,不同之处是其网络结构使用了迁移学习,输出为碱基与其他碱基形成氢键的可能性。而SPOT-RNA2的输入加上了经过LinearPartition[30]和RNAcmap[31]处理后的特征,且简化了网络。还有针对RNA结构的稳定性评判的计算方法,如RNA3DCNN[32]使用神经网络训练了基于知识的打分函数,可用于评价RNA三维结构的合理性。
与获得靶标三维结构相比,确定药物靶标结构中的活性位点同样重要。对于无已知活性化合物的药物靶标,需要通过可药性结合位点的预测方法来预测药物设计位点。传统的方法是使用探针分子,通过分子对接模拟的方法或靶标空腔(cavity)扫描等方法(如FTMap[33]和Fd-DCA[11]),寻找探针小分子集中结合的位点,作为预测的可药性位点。近日,一些基于深度学习算法的可药性位点的预测方法先后被报道。BiteNet[34]采用三维卷积神经网络通过对小分子-蛋白质复合物结构体系进行学习,从而建立药物设计位点的预测模型,该模型在GPCR、EGFR等体系都进行了应用测试,在精度与计算速度方面均表现出一定优势。PointSite[35]将蛋白质三维结构转换为点云,采用基于U-Net的子流形卷积(submanifold sparse convolution)方法进行分割,通过结合原子级表示和增强学习的手段,发展了可在原子水平预测蛋白质上小分子结合位点的算法。
当确定药物靶标的活性位点之后,便可采用分子对接的方法进行小分子化合物的虚拟筛选,即先导化合物的发现工作。传统的分子对接方法主要包括两个重要的模块,分子构象搜索与打分函数。打分函数是评价小分子与药物靶标亲和力的函数,是指导分子三维构象优化的目标函数,是决定分子对接方法效率的关键因素。然而,传统的分子对接打分函数基于力场和经验参数,在不同的药物靶标中很难同时取得良好的效果。因此,在先导化合物的发现过程中,仍然十分依赖药物化学家的经验判断。深度学习的方法直接从已有的复合物结构以及化合物-蛋白质的实验结合亲和力数据来建立预测药物-靶标结合亲和力模型。DeepDTA[36]使用了蛋白质残基序列,小分子结构SMILES(simplified molecular input line entry specification)编码,采用卷积神经网络,训练蛋白质、小分子与结合亲和力间的相关性的深度学习模型。另外,OnionNet[37]使用CNN网络,通过同时考虑蛋白质-小分子局部互作模式以及非局部互作模式,发展了小分子-蛋白质亲和力预测方法。在测试中,其结合亲和力预测值与实验值的相关性可达73%。除此之外,基于深度学习算法发展的小分子-蛋白质亲和力算法还有KDEEP[38]、RosENet[39]和DeepGS[40]等。鉴于篇幅所限,在此不一一赘述。
4.3 深度学习在分子生成中的应用
药物的化学合成是制约新药研发速度的关键过程。早期有机化学研究积累了大量的化合物数据以及合成路线信息,为设计新化合物并预测其可合成性提供了重要的学习信息。然而,对于真实的化学空间而言,人类目前所能成功合成的化学结构只是冰山一角。因此,设计具有新颖化学结构的化合物,并准确评价其可合成性的方法,将有力地推动新药研发的进程。针对上述问题,已有众多基于深度学习算法发展的用于类药性分子生成和有机分子的逆合成分析的方法被报道。对于分子生成方法,Méndez-Lucio等[41]使用生成式对抗网络[42](generative adversarial networks,GAN),在对SMILES编码时,使用了基于GRU的编码器和解码器,依赖于L1000数据库中小分子与基因表达差异的影响数据,建立了小分子与其对基因表达间的深度学习模型。该模型可根据某个基因敲除的基因表达图谱,生成具有特定调控基因表达功能的全新小分子。此外,ReLeaSE[43]使用了强化学习算法(其中网络部分使用到Stack-RNN)生成具有特定性质的分子。使用分子的图结构信息发展的分子生成方法,如DeepGraphMolGen[44]使用了基于图卷积神经网络的强化学习算法,生成具有类药性和可合成性的分子。
对于有机化学分子的逆合成分析,Liu等[45]使用了具有Attention机制的RNN,输入已有目标分子的SMILES和特定的化学反应类型,建立可预测反应产物的深度学习模型。此外,G2Gs[46]以一种图到图的框架,同样以上述信息作为学习数据,建立了预测模型来预测合成反应产物。
药物的吸收(absorption)、分布(distribution)、代谢(metabolism)、排泄(excretion)和毒性(toxicity)等性质,合称ADMET性质,决定了药物在临床研究中的成功率。其中,药物毒性的评价对于临床前研究至关重要。使用机器学习方法预测药物的ADMET性质已被广泛报道。2012年,唐赟课题组报道了基于机器学习技术发展的ADMET性质预测方法admetSAR[47],之后通过进一步丰富数据库,加入网络算法升级到admetSAR2.0版本[48]。此外,如Chemi-net[49]将分子转为图结构,并使用图卷积来预测ADMET性质。Wenzel等[50]使用全连接的深度神经网络发展了ADMET预测模型。ADMET预测模型是药物设计领域中机器学习算法最先介入后的研究成果。截至目前,基于AI算法而发展的预测模型也极其丰富,鉴于篇幅所限不再一一展开。
5 总结与展望
本文以介绍重要的深度学习算法为切入点,沿着药物研发主线,回顾了多种深度学习算法在药物研发初期的几个关键环节中的应用进展。深度学习算法在蛋白质结构预测与分子生成等诸多领域已经取得显著的成功。然而,仍有以下几个方面有待进一步探索:①数据集的数量和质量是改进深度学习的必要条件,如何有效产生、高效收集和准确处理数据将是进一步基于深度学习的药物设计关键问题;②如何设计和开发更高效且适宜于药物设计的深度学习算法,是进一步改进药物设计方法的另一关键;③如何针对药物设计中的不同问题,合理设计和提取学习特征,是弥补输入数据不足或网络学习能力不佳的关键手段。综上,鉴于深度学习算法在药物设计方法发展方面已经取得的成绩,以及生物医药行业技术的不断革新,我们对AI驱动的药物设计的未来充满信心,相信将有更多高效、高质量的药物设计方法被开发出来并推广应用。
猜你喜欢
肝博士(2022年3期)2022-06-30 02:48:48
北京航空航天大学学报(2021年9期)2021-11-02 08:24:26
海外星云(2021年9期)2021-10-14 07:26:10
军民两用技术与产品(2021年10期)2021-03-16 06:05:10
世界农药(2019年3期)2019-09-10 07:04:10
电子制作(2019年11期)2019-07-04 00:34:38
北京航空航天大学学报(2018年1期)2018-04-20 06:38:17
青苹果·教育研究版(2016年9期)2016-12-23 11:52:36
光学精密工程(2016年4期)2016-11-07 09:04:48
肿瘤影像学(2015年3期)2015-12-09 02:38:45
