
31例儿童脊髓性肌萎缩症临床与基因分析
2021-10-22 04:30:38陈瑜毅韩蕴丽黄诗琴
临床儿科杂志 2021年10期
陈瑜毅 韩蕴丽 李 杏 黄诗琴
1.广西医科大学第一附属医院(广西南宁 530021);2.广西壮族自治区妇幼保健院(广西南宁 530003)
脊髓性肌萎缩症(spinal muscular atrophy,SMA)是一种常染色体隐性遗传病,是由于脊髓前角运动神经元变性,导致肌肉无力和萎缩。临床表现为累及肢体近端的进行性、对称性的弛缓性瘫痪与肌萎缩。活产婴儿中SMA 的发生率是1/10000~1/6000,人群携带率为1/50[1],国内人群携带率约为1/42[2]。本文对31例SMA 患儿的临床资料进行回顾性分析,总结SMA 的临床和基因诊断,探讨SMN基因与临床表型的关系,以提高对SMA的认识,为临床干预和遗传咨询提供帮助。
1 对象与方法
1.1 研究对象
收集2014 年2 月至2019 年5 月在广西医科大学第一附属医院诊断的31例SMA患儿及其父母的临床资料以及基因分析结果。研究对象排除其他以肌无力为特征的相关疾病,包括各种类型的肌营养不良性疾病、脑性瘫痪迟缓型、先天性重症肌无力、先天性肌病、线粒体肌病、遗传性感觉运动神经病、脊髓延髓肌萎缩症等疾病以及临床资料不完整,未完善神经电生理检查及基因检测者。
1.2 方法
1.2.1 临床资料收集
收集患儿的起病年龄、性别、肌力、肌张力、运动里程碑发育等临床资料,总结临床及基因分析 结果。
1.2.2 遗传学检查
分别采集患儿及父母的外周静脉血各2 mL 行EDTA 抗凝处理。用QIAmp DNA Blood Mini Kit 提取基因组DNA,分离纯化、洗脱收集DNA,提取的DNA 片段进行分子量、浓度和纯度等检测。为避免DNA 分解先进行分装,置-20℃保存。采用多重连接依赖性探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)进行检测,严格按照SALSA MLPA probemix p060-B2 SMA试剂盒(荷兰 MRC-Holland 公司)说明进行DNA 标本变性、杂交、连接、PCR扩增、片段分离。应用Coffalyser.NET软件(MRC-Holland,www.mlpa.com-MLPA)进行数据分析。根据试剂盒说明,Ratio值为0时表明没有拷贝数,为纯合缺失;Ratio值为0.40~0.65时拷贝数为1,为杂合缺失;Ratio值为0.80~1.20时拷贝数为2,表示正常;Ratio值为1.30~1.65时拷贝数为3;Ratio值为1.75~2.15时拷贝数为4;Ratio值多于2个拷贝数的即为重复。
1.2.3 SMA分型
入选研究对象按2006 年世界神经病学联盟神经肌肉疾病研究组制订的临床诊断标准及分型标 准[3],主要依据起病年龄、病情进展情况及运动发育里程碑分为4 种主要常见类型。脊髓性肌萎缩症1型(Werdnig-Hoffman 病),婴儿型或严重型,一般在生后6个月内起病,生后即出现活动少,肌张力低,抬头不稳,可有宫内胎动减少史,通常在2 岁前死亡,是最严重的类型;脊髓性肌萎缩症2型(Dubowitz 病),为中间型,生后6~18 个月起病,患儿能独坐,但不能独立行走,可有关节挛缩和脊柱后凸,生存期一般在2 年以上,多数可活到成年;脊髓性肌萎缩症3型(Kugelberg-Welander病),为青少年型,多于生后18个月起病,能够独立行走,随着病情缓慢进展,逐渐出现肌无力症状,可伴有骨质疏松和脊柱侧弯,生存期可至成年;脊髓性肌萎缩症4型,晚发型,亦称成年型,通常在30岁或更晚起病,起病与进展较隐匿,是最轻的一种类型,运动功能可以达到终身正常行走且生存期与正常人无异。
1.3 统计学分析
采用SPSS 25.0 统计学软件进行数据分析。计数资料以百分比表示,样本率的比较采用χ2检验及Fisher精确检验,P<0.05提示差异有统计学意义。
2 结果
2.1 一般情况
31例SMA患儿中,男性20例、女性11例,男女比例1.8:1。6月龄内起病12例,占38.7%,年龄中位数为2.5个月;7~18月龄起病17例,占54.8%(17/31),年龄中位数为9 个月;18 月龄后起病2例,占6.5%,年龄中位数为48个月。1型SMA 12例(38.7%),2型17例(54.8%),3型2例(6.5%)。
患儿首发症状均为运动发育迟缓,13例(41.9%)肌张力低下,9例(29.0%)肌力下降,5例(16.1%)步态异常,4例(12.9%)生长发育迟缓。以近端肢体出现肌无力为主,下肢较上肢严重,腱反射减弱或消失;患儿均无感觉和认知变化。31例SMA 患儿的临床资料见表1。
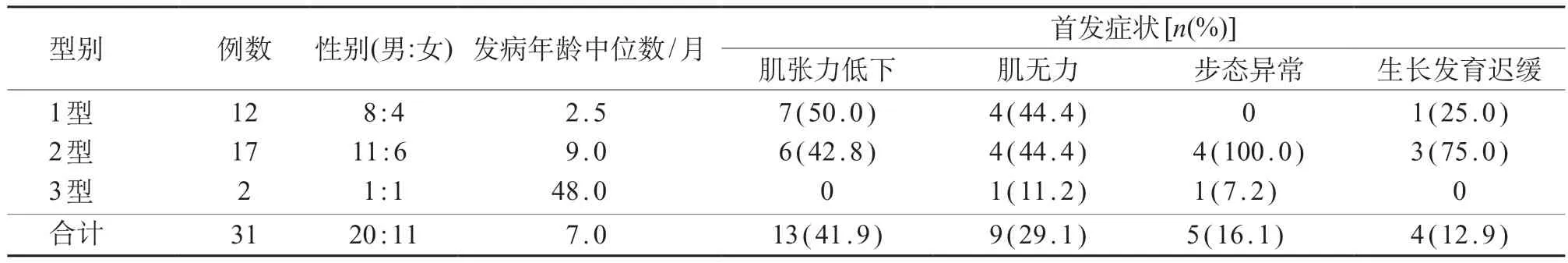
表1 31例SMA患儿临床资料
2.2 各型SMA患儿SMN1基因第7和/或8外显子缺失情况
31例临床诊断SMA患儿中,SMN1第7和8外显子纯合缺失29例,占93.5%,包括1型12例,2型15例,3型2例;仅有第7外显子缺失者2例,占6.5%(2/31),均为2型SMA。不同类型SMA 的临床表型与SMN 1基因缺失类型之间差异无统计学意义(P=0.415)。见 表2。
2.3 各型SMA患儿SMN2基因拷贝数变化情况
SMN 2基因拷贝数2 为12例(38.7%),其中1型SMA 10例(83.3%),2型SMA 2例(16.7%);拷贝数3 或4(重复)为19例(61.3%),其中1型SMA 2例(10.5%),2型SMA 15例(79.0%),3型SMA 2例(10.5%)。本组患儿中,2型和3型SMA患儿的SMN2基因拷贝数显著高于1型SMA 患儿,3型SMA 患儿的SMN 2基因拷贝数明显高于2型SMA 患儿,不同SMA 临床表型与SMN 2拷贝数差异有统计学意义(P<0.05)。见表3。
2.4 SMA患儿父母亲SMN1、SMN2基因拷贝数情况
30例(96.8%)患儿的父母亲均明确诊断为SMN 1基因杂合缺失;1例(3.2%)患儿父亲明确诊断为SMN 1杂合缺失,母亲SMN 1基因未检测到纯合或杂合缺失。即有61例(98.4%)双亲携带1个SMN 1基因拷贝数,提示为SMA 致病基因携带者。包括SMN 1缺失(SMN 1:SMN 2=1:1,16例;SMN 1:SMN 2=1:2,20例)产生的携带者36例,占59.1%;SMN1转换为SMN2(SMN1:SMN2=1:3,13例;SMN 1:SMN 2=1:4,12例)产生的携带者25例,占40.9%。1例(1.6%)患儿母亲携带2 个SMN 1和SMN2基因拷贝数。见表4。
3 讨论
根据2006 年世界神经病学联盟神经肌肉疾病研究组制订的SMA 诊断标准及分型标准[3],依据起病年龄、病情进展情况及运动发育里程碑,可将SMA分为4型,分别为SMA-1型、SMA-2型、SMA-3型、SMA-4型。因SMA-4型在成人期起病,根据年龄划分,儿童时期SMA主要为SMA-1型(婴儿型)、SMA-2型(中间型)及SMA-3型(青少年型)。在临床上,各型SMA 严重程度差别大,起病年龄可从新生儿期至成人期,运动发育障碍从不能抬头至可独立行走,存活期可到婴幼儿至成年不等。本组31例SMA 患儿中,SMA-1型12例(38.7%),2型SMA 17例(54.8%),3型SMA 2例(6.5%)。首发症状包括肌张力低下、肌力下降、步态异常、生长发育迟缓等。各类型间在首发临床表现中有重叠现象,因此在临床分型中,还需观察有无其他临床表现以及在病情进展过程中患儿运动功能获得情况作为分型依据。
近年来,随着分子遗传学研究迅速发展,SMA患儿可以通过基因检测得到明确诊断。运动神经元存活1基因(survival motor neuronal gene 1,SMN1)于1995 年由法国学者Lefebvre 等[4]首次克隆成功,定位于5 q 11.2-13.3 区域内,该基因缺失是脊髓性肌萎缩症的致病基础,其突变导致运动神经元存活蛋白(survival motor neuron,SMN)表达水平降低或功能丧失。体内多数细胞都能耐受低水平的SMN蛋白,但脊髓前角运动神经元不能耐受低水平SMN 蛋白,最终导致疾病的发生[5]。人类SMN基因有两个高度同源的拷贝,即SMN1和SMN2;两者具有5个核苷酸的差异,但在编码区仅有1个核苷酸的差异(c.840C>T),导致SMN 2基因转录剪切中发生跳跃,其缺少外显子7 编码的核心功能区,只表达为全长SMN蛋白(FL-SMN)的10%~20%,且大部分编码1个不具生物功能、极不稳定且易分解的截短蛋白(∆7-SMN)[6]。因此,SMN2基因只有在SMN1基因缺失情况下才起到剂量补偿的作用。
在国内人群中,约95%的SMA 患儿存在SMN 1基因第7外显子和/或第8外显子的纯合缺失[7],且大多数同时缺失外显子7和外显子8,只有少部分患儿仅缺失外显子7。本研究采用MLPA技术进行基因检测。SMN1外显子7和8纯合缺失者有29例,占93.5%,包括1型SMA 12例(41.4%),2型SMA 15例(51.7%),3型SMA 2例(6.9%);仅外显子7 缺失者有2例(全部为2型SMA),占6.5%。本组SMA患儿中未发现只有外显子8缺失的病例。其原因为SMN基因的终止密码子位于外显子7 的末端附近,外显子8 并不编码氨基酸。SMA患儿正是由于外显子7编码的核心功能区缺失和SMN 1基因重要功能丧失所致。本研究表明,不同SMA 临床表型与SMN 1基因缺失类型之间差异无统计学意义,因此,SMN 1基因缺失的测定在各型SMA患儿中具有相同的临床价值。该结论与Fang等[8]的报道一致。
本研究中,2型和3型SMA 患儿的SMN 2基因拷贝数显著高于1型患儿,3型SMA 患儿SMN 2基因拷贝数明显高于2型患儿,表明不同SMA 临床表型与SMN 2拷贝数有显著差异,提示SMN 2基因拷贝数与疾病的临床表型严重程度相关。SMN 2基因通常被认为是脊髓性肌萎缩的修饰基因。当SMN1基因缺失时,SMN 2基因通过转换部分补偿了表达全长SMN 蛋白的功能。该结果与Qu等[9]报道一致。另外,相同拷贝数的SMN 2基因可存在于各类型的SMA 患儿中。本研究中SMN2基因拷贝数2为12例,其中1型SMA 10例,2型SMA 2例;拷贝数3或4(重复)为19例,其中1型SMA 2例,2型SMA 15例,3型SMA 2例。因此,虽然SMN2基因的拷贝数可解释大多数SMA的表型变化,但应该注意的是,SMN 2基因的拷贝数并不能完全预测表型的严重程度[10]。这是因为在SMA患儿中还有其他的修饰基因,如神经元凋亡抑制蛋白(neuronal apoptosis inhibitory protein,NAIP)基因、基本转录因子2p44 (basal transcription factor 2p44,BTF2p44)基因和H4F5基因。另外在王旻晋等[11]研究中发现,SMN 2在3型SMA 中有更多的拷贝数5 或6。本研究中没有发现更多的SMN2拷贝数,考虑与样本数量少有关。
另一项研究显示,约5%的SMA患儿存在SMN1基因复合杂合突变[12],即存在一个SMN 1基因缺失,并伴有另一个SMN 1基因内的点突变。如果患儿临床表型与SMA 一致,但未能检测出SMN 1第7 外显子纯合缺失,有必要再次进行临床评估,另外应考虑存在SMN1基因点突变的可能性,并需进一步的基因测序证实,从而确定是否为复合杂合突变的SMA 患儿。2个SMN1基因均发生点突变者极少,国内尚无报道[10]。本研究中31例SMA患儿经MLPA检测均为纯合缺失,未发现复合杂合突变。这与纳入的病例数少 有关。
本研究中有30例(96.8%)患儿父母亲明确为SMN 1基因杂合缺失,1例(3.2%)患儿父亲明确诊断为SMN 1杂合缺失,母亲SMN 1基因未检测到纯合缺失或杂合缺失。即有98.4%的父/母亲携带1个拷贝数的SMN 1基因,表明他们均为SMA 致病基因携带者,其中SMN 1缺失(SMN 1:SMN 2=1:1 和SMN 1:SMN 2=1:2)引起的占59.1%,SMN 1转换为SMN 2(SMN 1:SMN 2=1:3 和SMN 1:SMN 2=1:4)引起的占40.9%。1例(1.6%)患儿母亲携带2 个拷贝数的SMN 1基因。推测可能是SMN 1基因“2+0”型(即含2 个拷贝数的SMN 1基因位于同一条染色体上,另一条同源染色体缺失SMN 1基因)、“1+1 D”型(即含1 个拷贝数的SMN 1基因位于一条染色体上,另一条同源染色体发生点突变)或无义突变等所致。龚波等[13]发现SMA 携带者中有1 个拷贝数的SMN 1和0 个拷贝数的SMN 2个体。但在本研究中并没有发现该比值的携带者,考虑与研究病例数少 有关。
本研究仅应用MLPA技术进行基因检测,该方法未能检测到上述突变。这也需要通过进一步连锁分析或基因测序来确认。应该对此类父母进行产前遗传咨询。因此产前诊断是减少SMA 患儿出生、降低SMA发生率的一种手段,是减轻社会和家庭负担最有效的途径[14]。在部分国家中,SMA检测方法已被纳入孕前筛查[15-17],特别是对防止先证者家庭再次生育SMA子女具有重要意义。
本研究受检测条件限制,未对除SMN2以外的修饰基因及可能发生SMN1复合杂合变异类型进行更深入研究,有待今后联合其他检测方法进一步研究。
猜你喜欢
河北医学(2021年10期)2021-10-27 00:37:14
种子(2021年3期)2021-04-12 01:42:22
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:48
西安交通大学学报(医学版)(2015年2期)2015-02-28 17:59:20
发明与创新(2015年25期)2015-02-27 10:39:16
医学综述(2014年24期)2014-03-08 07:07:24
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
山西农业大学学报(自然科学版)(2011年3期)2011-04-25 10:17:50
