
急性脑干综合征起病的视神经脊髓炎谱系疾病1例
2021-10-19 02:39宋阳江沛
巴楚医学 2021年3期
宋 阳 江 沛
(宜都市第一人民医院 神经内科, 湖北 宜昌 443300)
视神经脊髓炎(neuromyelitis optica,NMO)也称Devic病,是一种特发的严重中枢神经系统脱髓鞘疾病,主要影响视神经和脊髓[1]。高特异性血清自身抗体标记物(neuromyelitis optica immunoglobulin G,NMO-IgG)的存在可以用来区分NMO和多发性硬化症,并有助于确定NMO疾病谱,而NMO-IgG主要与Aquaporin 4反应[2]。东亚人群中,NMO谱系疾病(neuromyelitis optica spectrum disorders,NMOSD)的患病率约为3.5/10万,而在Aquaporin 4抗体阳性的病例中,女性明显占优势(高达90%),而且大多数病例为成年人(40岁左右)[3]。临床症状最初表现为脊髓炎的占48%,视神经炎的占42%,最后区综合征的占10%,脑干/间脑/脑症状的占14%,同时出现视神经炎和脊髓炎的占4%[4]。现报道1例以急性脑干综合征起病的NMOSD。
1 病史摘要(扫码观看视频,D009471-1)
患者,女,32岁,因“头痛7 d,头晕、复视3 d”于2021年4月27日入院。患者于7 d前受凉后突然出现头痛,呈持续性隐痛,并逐渐加重,3 d前出现头晕,伴有复视及阵发性口周麻木不适,无恶心、呕吐,无肢体乏力、麻木,无胸痛、心悸,无腹痛、腹泻,门诊以“多发性硬化?”收住我科。既往有类似发作史,予以激素治疗后好转。查体:T 36.2℃,HR 80次/min,BP 120/70 mmHg,R 19次/min,心肺腹部体格检查无异常。专科体检:双侧瞳孔等大等圆,直径3 mm,对光反射灵敏,双眼向右侧偏斜,右眼向上、向内活动受限,左眼向外、向上活动受限,可见水平粗大眼球震颤,皱额正常,鼻唇沟对称,无构音障碍,伸舌居中,肌力、肌张力正常,Romberg征不配合,双侧Babinski征(-),颈软,Kernig征(-)。辅助检查:血常规:中性粒细胞比率 81.0%↑,血红蛋白 99 g/L↓;肝功能、血糖、血脂、生化、心肌酶谱、凝血功能、D-二聚体正常。头颈部MRI示(见图1):脑桥背侧及中脑导水管周围异常信号影,T1加权像(T1-weighted images,T1WI)等信号、T2加权像(T2-weighted images,T2WI)稍高信号、T2液体衰减反转恢复序列(T2 fluid attenuated inversion recovery,T2-FLAIR)高信号影,考虑脱髓鞘改变可能;考虑双侧额叶及右顶叶少许缺血灶;右侧筛窦炎,双侧下鼻甲肥大;颈椎退行性变;C4/5、C5/6椎间盘突出;下丘脑内侧缘、侧脑室前角及第三脑室室管膜下异常信号影。行腰椎穿刺脑脊液检查,测压150 mmH2O;脑脊液(cerebrospinal fluid,CSF)常规:白细胞计数6×106/L,单个核白细胞百分比 100%;CSF生化检查:脑脊液蛋白0.282 g/L,葡萄糖3.5 mmol/L,腺苷脱氨酶0.2 U/L,乳酸脱氢酶24 U/L↓,氯123 mmol/L↑;脑脊液抗酸染色、革兰染色、真菌涂片、墨汁染色、培养无异常。外送查脑脊液寡克隆带检查示:IgA值升高,经脑脊液和血清等电聚焦电泳分析,均未见IgG寡克隆条带。血液中枢神经脱髓鞘抗体检测示:抗血清水通道蛋白4免疫球蛋白G抗体(aquaporin 4-immunoglobulin G,AQP4-IgG) 1∶100。
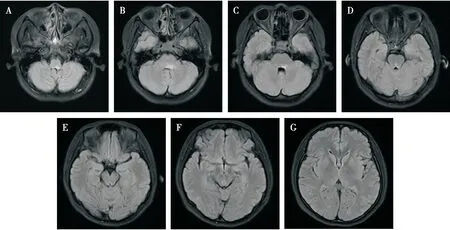
注:A、B:延髓背侧T2-FLAIR高信号;C:脑桥背侧T2-FLAIR高信号;D、E:中脑导水管T2-FLAIR高信号;F:下丘脑内侧缘T2-FLAIR高信号;G:侧脑室前角及第三脑室室管膜下T2-FLAIR高信号图1 该患者头颅MRI检查结果
综合患者病史及辅助检查:患者急性起病,表现为头痛、头晕、复视及口周麻木不适;查体:双眼向右侧凝视、可见眼球震颤及眼肌麻痹;头颅MRI主要表现为侧脑室前角、第三脑室、中脑导水管及第四脑室周围异常信号;结合外送血清抗AQP4抗体阳性,考虑患者为急性脑干综合征起病的NMOSD。入院d 2起予以甲泼尼龙琥珀酸钠1 g,连用3 d后序贯减量治疗,辅以营养神经、抑酸、补钙等对症治疗,患者症状明显好转。出院后口服泼尼松片 60 mg,1次/d(每周减5 mg)。2周后电话随访,患者诉头痛、头晕、复视及口周麻木症状消失。
2 讨论
NMO是一种罕见的自身免疫性炎症和脱髓鞘疾病,以中枢神经系统为靶点。NMOSD是一种新修订的命名方法,最近的研究提出了新的诊断标准,包括AQP4-IgG的血清学检测。伴有AQP4-IgG阳性的NMOSD诊断标准要求至少有1个与视神经炎、急性脊髓炎、最后区综合征、急性脑干综合征、间脑综合征或脑综合征相关的核心临床特征或MRI表现。对于没有AQP4-IgG的NMOSD诊断,需要更全面的临床资料和额外的神经影像学发现[5]。
在NMOSD中,脑干病变最常见的表现为呕吐和顽固性打嗝,其次为眼球运动障碍(复视及眼球震颤)、瘙痒,其他少见症状有听力障碍、面瘫、三叉神经痛、眩晕或前庭性共济失调、构音障碍、吞咽困难、脑神经XII、IX麻痹等其他表现。这些症状多数是可逆的,但也可能有后遗症,尤其是听力障碍和眼球运动障碍的患者[6]。与以视神经脊髓受累为初始症状的NMOSD相比,脑干病变的患者起病年龄更年轻,血清AQP4抗体滴度较低,临床预后更好[7]。
NMOSD的特征性MRI表现为T2-FlAIR高信号,通常不对称分布于室管周围区域,位于侧脑室、第三脑室和第四脑室管室内衬,特别是靠近中脑导水管。胼胝体室管膜表面、间脑区和脑干也被认为是NMOSD脑损伤的典型部位。 Cao等[8]研究显示,在270例患者中,NMOSD最常见的表现为非特异性白质改变(40%),17.7%的患者出现NMOSD特征性病变,近1/3 (36.3%)患者MRI表现正常,6%患者表现为多发性硬化(multiple sclerosis,MS)样病变。对于7.0 T MRI在NMOSD中的应用也有报道,但主要是通过对中央静脉、铁沉积、皮质病变的磁共振成像分析以鉴别MS[9]。
大剂量静脉注射甲基强的松龙是NMOSD的标准治疗方法。对皮质类固醇治疗疗效不显著的患者,血浆置换或免疫吸附的升级治疗是必要的,甚至可以作为一线治疗[10]。硫唑嘌呤(azathioprine,AZA)、霉酚酸酯(mycophenolate mofetil,MMF)(非选择性抑制免疫细胞的快速分裂)或利妥昔单抗(rituximab,RTX)(一种针对B细胞表面CD20的单克隆抗体,以诱导B细胞衰竭)可作为预防NMOSD复发一线药物,这些药物都能降低复发率,并可能稳定甚至改善神经功能障碍[11]。低剂量糖皮质激素在NMOSD中的应用尚未进行系统研究,然而其经常被用作维持治疗、单用或添加常规免疫抑制剂、或在复发后慢慢减少使用[12]。最近,FDA批准了三种用于治疗AQP4抗体阳性NMOSD的单克隆抗体,即eculizumab、inebilizumab和satralizumab,但关于这些新药的长期安全性和有效性仍有待研究[13]。
本例NMOSD的特殊之处在于以脑干受累起病,而非常见的视神经炎、脊髓炎为主要表现形式,接诊初期因贫血及头部MRI表现一度无法排除Wernicke脑病,但患者既往类似发作病史及激素治疗有效提供了新的诊疗思路。AQP4-IgG抗体阳性证实了为脑干病变的NMOSD,然而对于初发以脑干受累的NMOSD、甚至AQP4抗体阴性的该类患者,极易造成误诊,临床医师应引起重视,注意鉴别。通过电话随访,患者经治疗后临床症状完全消失。与以视神经脊髓受累为初始症状的NMOSD相比,脑干病变的患者临床预后更好。
猜你喜欢
重庆医学(2022年18期)2022-10-08
健康博览(2022年5期)2022-05-24
中国典型病例大全(2022年12期)2022-05-13
中国听力语言康复科学杂志(2021年6期)2021-12-21
中国听力语言康复科学杂志(2021年6期)2021-12-21
罕少疾病杂志(2021年5期)2021-09-27
中华养生保健(2021年18期)2021-02-13
科教新报(2019年26期)2019-09-10
中国医药导报(2011年27期)2011-12-31
祝您健康(1998年9期)1998-12-25
