
团簇Mo3S4的成键及电子性质
2021-10-15 06:05朱依文方志刚候欠欠王思怡
辽宁科技大学学报 2021年3期
朱依文,方志刚,许 友,候欠欠,王思怡
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
团簇是存在于原子、分子与宏观固体材料之间的一种物质,可以作为均相催化剂和多相催化剂的有效模型[1-4]。二元团簇Mo-S体系以其独特的光催化性[5]、电化学性等优异性能,受到广泛关注。二元团簇Mo-S体系由金属元素钼和非金属元素硫组成。金属钼导电性能优异,热胀系数小,被广泛应用于电子管、整流器等电子仪器中。非金属硫的化合物在矿物冶炼时往往会生成二氧化硫,造成严重的大气污染。为解决这一问题,目前石油加工主要采用过渡金属硫化物催化剂进行脱硫。迄今为止,已有二十余种Mo-S体系化合物用于矿物燃料的加氢脱硫工业,成为铂族金属的低成本替代品,作为加氢和析氢反应催化剂[6-8]。在Mo-S体系中,团簇Mo3S4被认为是一种很有前途的仿生电催化剂,对H2的活化过程起显著的促进作用。团簇Mo3S4具有超导电性,使其在电子学领域有巨大的应用前景。与此同时立方烷型团簇Mo3S4氢化物催化硝基芳烃在医学上亦发挥着十分重要的作用[9]。团簇Mo3S4具有众多优点,但目前以宏观实验角度的研究甚少,本文从微观角度对团簇Mo3S4各优化构型的成键以及电子性质进行分析,以期为今后研究团簇Mo3S4提供理论基础。
1 团簇Mo3S4的优化构型
以拓扑学原理为基础,根据密度泛函理论[10],在B3LYP水平上进行研究,设计出团簇Mo3S4所有可能存在的构型,并对其一、三重态进行全参数优化计算,将所有含虚频和相同的构型进行排除,最终得到了8种优化构型。对Mo、S原子采用Hay[11]等人的18-eECP双ξ基组,并对S加极化函数ξs.d=0.55[12]。所有运算过程均在启天M4390上的Gaussian09程序中运行。
8种优化构型如图1所示。其中单、三重态各为4种,所有构型均为立体构型。右上角括号内的数字1和3代表构型所属的重态。基于空间结构的差异将团簇Mo3S4所有优化构型分为三大类:五棱双锥型(1(1)、2(1))、戴帽四棱双锥型(2(3)、3(1)、3(3)、4(3))和戴帽三角双锥型(1(3)、4(1))。以具有最低能量值的构型1(1)作为参考构型,将其能量值设为0 kJ/mol,计算出其余构型的相对能量,按各构型能量由小到大排序:1(1)<1(3)<2(1)<2(3)<3(3)<4(3)<3(1)<4(1)。
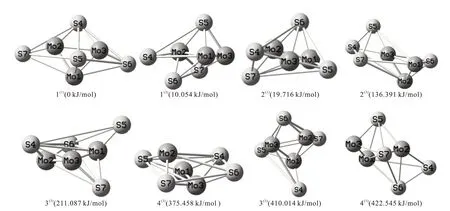
图1 团簇Mo3S4的8种优化构型Fig.1 Eight optimized configurations of cluster Mo3S4
2 团簇Mo3S4的成键
键长是共价键的重要参数之一,通过对键长的分析,研究团簇Mo3S4的成键性质。键长指用于成键的两个原子核间的距离,一般来说,键的数目越多,键长越短,原子距离越小,结合越牢固,所形成的化学键作用越强越不易断裂。但由于受到空间位阻效应、孤对电子的排斥力等多方面因素的影响,即使在同一个分子中,相同的两个原子形成的化学键键长也存在一定的差异,因此,通过分析键长的平均值来分析该键的成键强度。
依据分子轨道法理论,由两个符号相同的原子轨道线性组合所形成的新轨道相较于原轨道的能量更低,为成键轨道,成键轨道上电子形成的化学键更加稳定;而两个符号相反的原子轨道线性组合形成的反键轨道上的电子使轨道能量升高,不利于形成稳定的化学键。键级=(成键电子数-反键电子数)/2,可通过键级判断相邻两个原子的成键强度。键级越大,说明形成的共价键强度越大,所形成的化学键就越稳定。
表1和表2分别列出了团簇Mo3S4的8种优化构型中Mo-Mo键、S-S键及Mo-S键的平均键长和平均键级数据。各个构型中S-S键的键长最大,说明S-S键的共价性弱,稳定性弱。而Mo-Mo键的键长最小,说明Mo-Mo键的共价性强,稳定性强。构型3(1)的三种键的键长均出现较大波动,其余各个构型的Mo-Mo键的平均键长波动均较为平缓,说明除构型3(1)外的其余构型Mo-Mo键均处于相对稳定的状态。前六个构型中的S-S键和Mo-S键的键长变化趋势恰好相反,说明这两者可能存在拮抗作用。
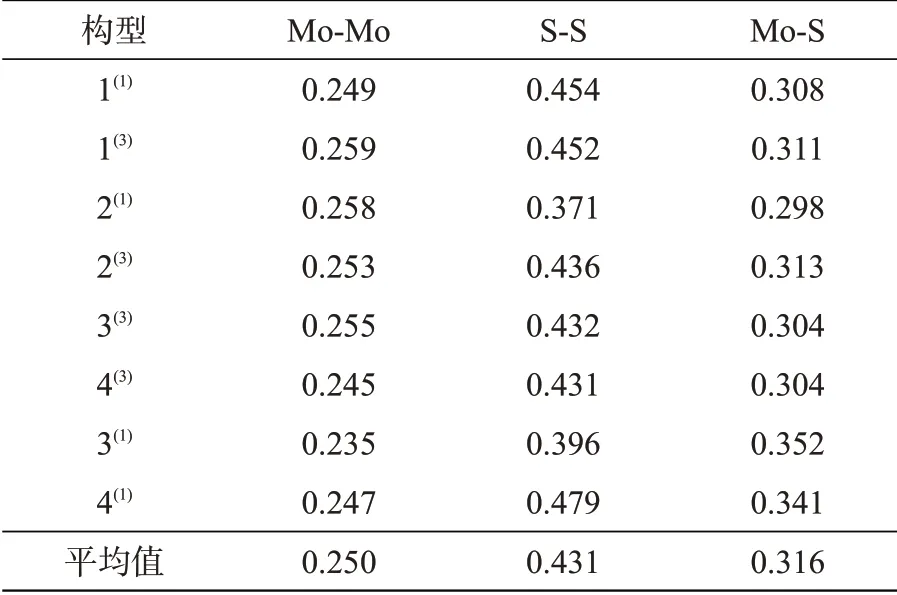
表1 团簇Mo3S4的8种优化构型平均键长,nmTab.1 Average bond lengths of 8 optimized configurations of cluster Mo3S4,nm
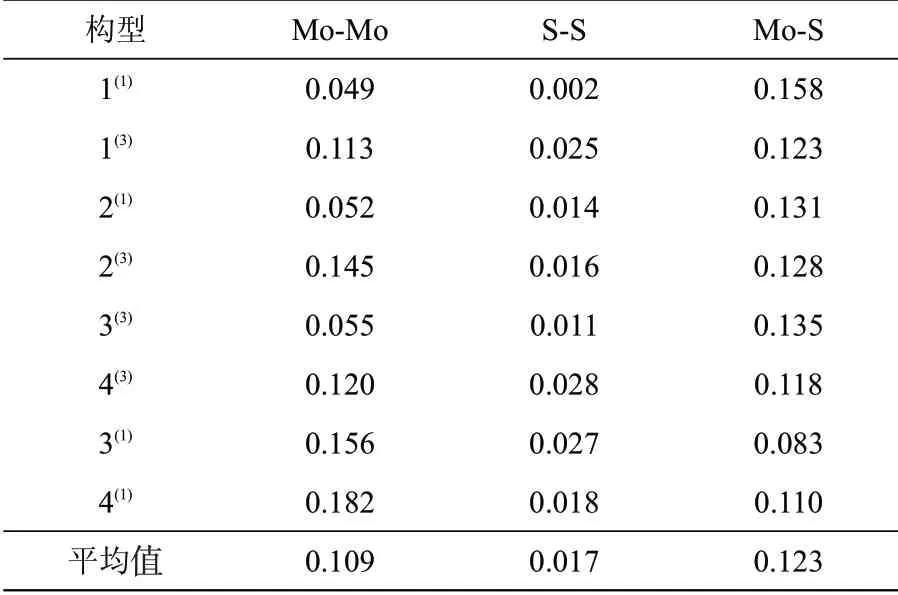
表2 团簇Mo3S4的8种优化构型平均键级Tab.2 Average bond orders of 8 optimized configurations of Mo3S4
Mo-Mo、S-S、Mo-S键级的平均值均大于0,说明成键轨道的能量更高。S-S键的键级远小于另外两种键,说明该键对构型的稳定性起较小的作用。Mo-S键的平均键级最大,说明Mo-S键更易形成稳定的键。除构型4(1)外,S-S键和Mo-S键的平均键级亦存在此消彼长的变化趋势,说明S-S键和Mo-S键存在一定的拮抗作用。结合键长和键级的数据分析,S-S键的键长最长且键级最小这说明团簇Mo3S4中S-S键成为成键作用的主要贡献者可能性极小。Mo-Mo键与Mo-S键的键长均小于S-S键,但Mo-S键的平均键级最大,说明Mo-S键是团簇Mo3S4稳定性的主要贡献者。
表3为团簇Mo3S4优化构型中各键键级占总键级的百分比。金属-非金属键键级占比(Mo-S)>金属-金属键键级占比(Mo-Mo)>非金属-非金属键键级占比(S-S),表明Mo-S键为团簇Mo3S4优化构型成键作用的主要贡献者。
3 团簇Mo3S4的电子性质
3.1 电荷量
通过分析团簇Mo3S4各原子之间得失电子即电荷转移的数量,进一步分析Mo、S原子间的相互作用。团簇Mo3S4各原子的电荷量值如表4所示,当电荷量为正值时说明该原子失去电子,为团簇Mo3S4提供电子;当电荷量为负值时说明该原子得到电子,即成为团簇中接受电子的一方。各原子所带电荷量绝对值越大,越能激发电场。
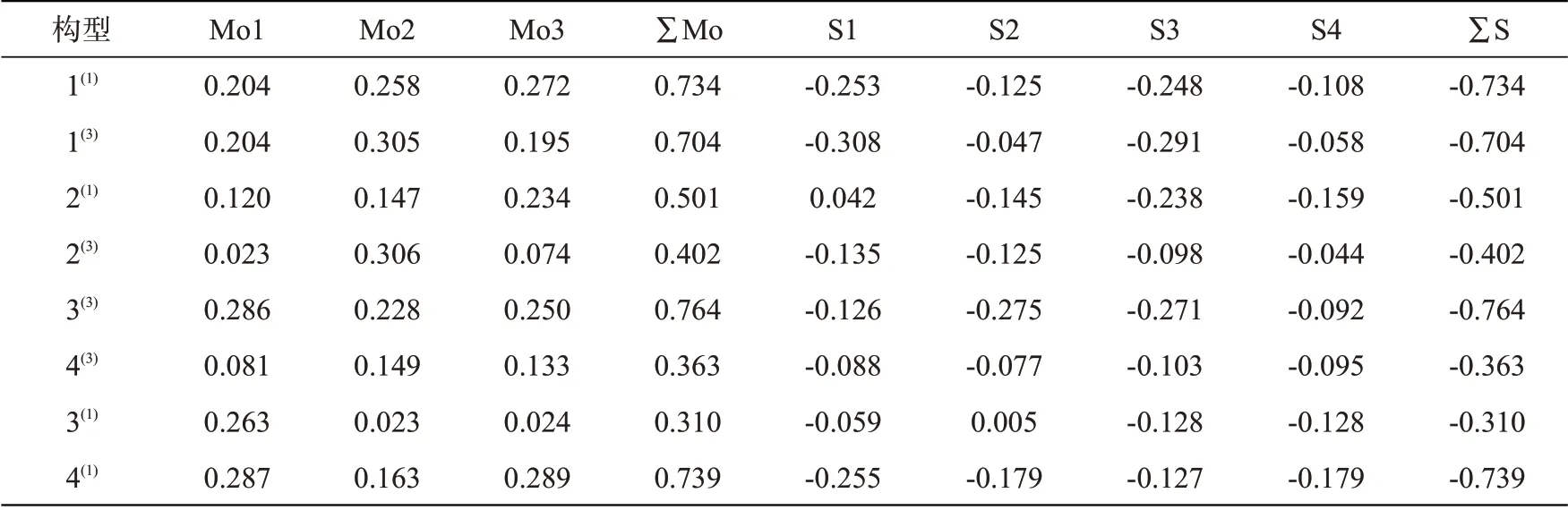
表4 团簇Mo3S4中各个原子的电荷量Tab.4 Charge of each atom in cluster Mo3S4
∑Mo和∑S表示相应原子的带电量之和,∑Mo+∑S=0,说明该团簇呈现出电中性。所有Mo原子的电荷量均为正值,所有S原子的电荷量均为负值,这表明团簇Mo3S4中电子均由Mo原子流向S原子。这是由于S元素的电负性(2.5)强于Mo元素的电负性(1.8),S原子吸引成键电子的能力更强。
根据表4数据做出如图2所示的各优化构型原子所带电荷变化趋势图。Mo原子与S原子的电荷变化趋势完全相反,关于x轴对称。电荷量越大说明电子流动性越强,各优化构型电子流动性排序:3(1)<4(3)<2(3)<2(1)<1(3)<1(1)<4(1)<3(3)。
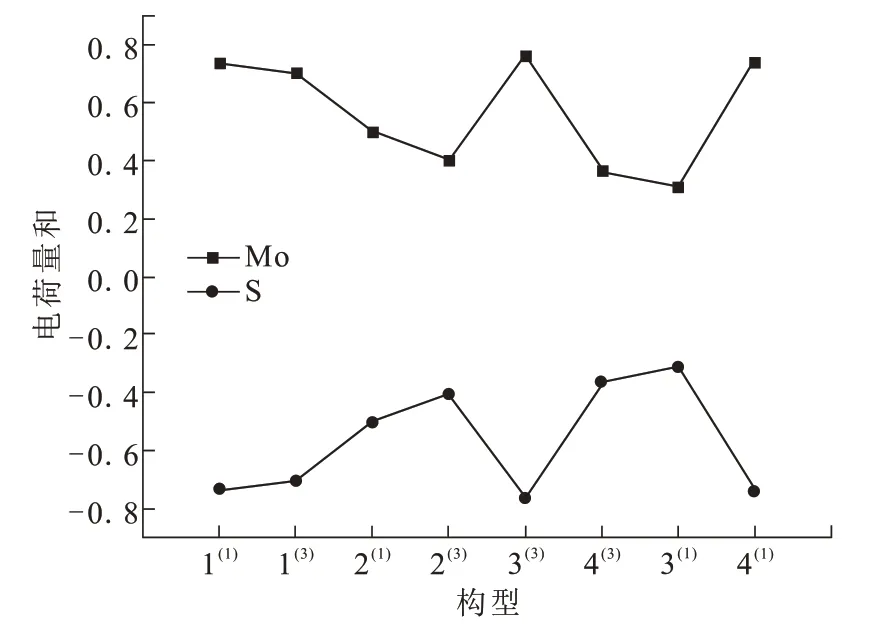
图2 各优化构型原子所带电荷量和Fig.2 Sum of charges of atoms in each optimized configuration
3.2 团簇Mo3S4稳定性规律分析
单重态分子的电子自旋成对,净自旋为0,而三重态分子中含有两个自旋不配对的电子。根据洪特规则,处于分立轨道上的非成对电子,平行自旋比成对自旋更稳定。因此,三重态能级总是比相应的单重态更低,也更为稳定。
团簇Mo3S4的4种三重态优化构型电子自旋密度分布如图3所示。图中浅色表示α电子,深色表示β电子。对各优化构型的稳定性进行分类讨论。第一类,对称性较好的构型1(3)和4(3),呈对角线对称;构型1(3)周围均匀围绕着顺磁场方向的ɑ电子,同时ɑ电子与β电子重叠程度略大于构型4(3),说明构型1(3)的稳定性优于构型4(3)。第二类,对称性不明显的构型2(3)和3(3),ɑ与β电子重叠程度不均匀且重叠面积较小,说明第二类构型的稳定性不如第一类构型。综上所述,构型1(3)的电子自旋密度分布对称性与重叠程度均最好,故其构型最为稳定。
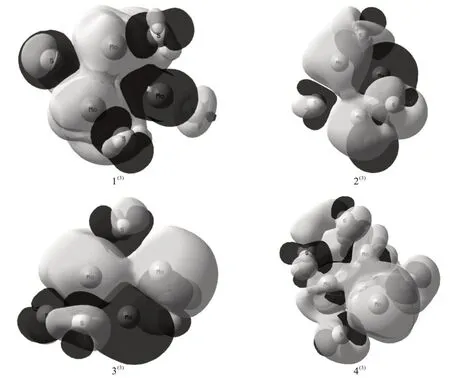
图3 团簇Mo3S4的4种三重态优化构型的电子自旋密度分布图Fig.3 Electron spin density distributions of four triplet optimized configurations of cluster Mo3S4
4 结论
团簇Mo3S4有单、三重态各四种的稳定构型,分别为五棱双锥、戴帽四棱双锥、戴帽三角双锥。根据各键级占总比的平均值进行分析,发现金属-非金属键键级占比(Mo-S)>金属-金属键键级占比(Mo-Mo)>非金属-非金属键键级占比(S-S)。Mo-S键为团簇Mo3S4优化构型的成键作用的主要贡献者。团簇Mo3S4中电子均由Mo原子流向S原子。团簇呈电中性。构型的电子流动性排序:3(1)<4(3)<2(3)<2(1)<1(3)<1(1)<4(1)<3(3)。构型1(3)的电子自旋密度分布对称性与重叠程度均优于其他三重态构型,说明构型1(3)的稳定性最好。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
天津理工大学学报(2021年2期)2021-06-03
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
新高考·高一物理(2016年7期)2017-01-23
新高考·高一物理(2015年6期)2015-09-28
