
UPLC-MS/MS测定人血浆中诺氟沙星的含量及其应用
2021-10-09 08:15:52刘海姣廖音娟唐智长沙市第四医院长沙40006长沙都正生物科技有限责任公司长沙40000
中南药学 2021年9期
刘海姣,廖音娟,唐智(.长沙市第四医院,长沙 40006;2.长沙都正生物科技有限责任公司,长沙 40000)
诺氟沙星是一种广谱抗菌剂,已在多个国家使用,可有效治疗人类和动物的感染,用于治疗尿路感染、呼吸道感染、胃肠道感染、伤口感染等[1-5]。目前,有研究报道了海水、生物体液、食物以及人血浆中诺氟沙星的测定方法[4,6-8],常用的技术有紫外或荧光检测的高效液相色谱法、毛细管电泳法、分光光度法、薄层色谱法、微生物法等[9]。Maia 等[10]使用基于HLB 柱的固相萃取(SPE)进行液相色谱-串联质谱(LC-MS/MS)定量污水中7 种不同的氟喹诺酮类抗菌药物,Ziarrusta 等[11]使用液相色谱串联质谱法测定鱼组织、生物体液和环境水中的氟喹诺酮类药物。Helmy 等[12]用高效液相测定血浆中诺氟沙星的含量,Amjadi 等[13]使用硅掺杂碳点、铁(Ⅱ)和K2S2O8组成的化学发光系统测定人血浆中的诺氟沙星,有研究采用LC-MS/MS 法并使用环丙沙星作为内标定量测定人血浆中诺氟沙星的浓度[14],也有报道采用超高效液相色谱-串联质谱(UPLCMS/MS)法测定血浆中诺氟沙星的浓度[15],但所需分析时间普遍较长(4 min 以上),未完善对方法学的考察,如未进行样品溶血基质效应、高血脂基质效应及全血稳定性的考察,这些因素均可影响到样本测试结果的准确性和可靠性。因此建立一种准确、可靠的检测人血浆样品中诺氟沙星浓度的分析方法至关重要。
本试验采用UPLC-MS/MS 法,以同位素为内标测定人血浆样品中诺氟沙星的浓度,检测过程更加简单、快速,极大地缩减了分析时间和项目实施成本,可以更好地应用于诺氟沙星人体生物等效性研究。
1 材料
1.1 仪器
UPLC I-Class、Xevo TQD 质谱仪,配有电喷雾离子源,UNIFI 数据处理软件(美国Waters 公司);MSA6.6S-CE 电子天平(Sartorius 公司);MilliQ Direct 8 超纯水仪(Merck Millipore公司);ST16R 离心机(Thermo Fisher Thermo 公司);Pipetman NEO 和Pipet-Lite XLS+移液器(Gilson 和RAININ 公司);XW-80A 涡旋混合器(宁波新芝);Talboys 数显多管式涡旋震荡器(Troemner 公司);DTA-27 超声波清洗器(鼎泰恒盛);GZX-9140MBE 电热鼓风干燥箱(上海博迅);MLTS1368 医用低温冰箱(Thermo 公司)。
1.2 试药
诺氟沙星(中国食品药品检定研究院,批号:13450-201206,纯度:99.5%);内标诺氟沙星-d8(批号:12-ZCA-49-4,纯度:97%,TRC)。甲醇、乙腈(色谱纯,Merck 公司);甲酸(FA,色谱纯,ACS);水为试验室超纯水仪自制水。
2 方法
2.1 色谱条件
色谱柱:Waters ACQUITY UPLC HSS T3(2.1 mm×50 mm,1.8 µm);流动相:0.2%甲酸水-乙腈=85∶15(V/V);流速:0.4 mL·min-1;运行时间:1.5 min;进样量:10 μL;柱温:40 ℃;自动进样器温度:20 ℃。
2.2 质谱条件
采用电喷雾离子源(ESI),正离子模式,MRM 监测,离子源温度为150℃,毛细管电压为0.5 kV,脱溶剂气流为1000 L·h-1,温度为500℃,诺氟沙星与内标诺氟沙星-d8 的离子对分别为m/z320.11 →m/z276.13 和m/z328.17 →m/z284.20。其中诺氟沙星的锥孔电压为40 V,碰撞能为14 eV,驻留时间为0.08 s;诺氟沙星-d8 锥孔电压为40 V,碰撞能为16 eV,驻留时间为0.08 s。
2.3 标准工作液及质控工作液的配制
精密称取诺氟沙星对照品和诺氟沙星-d8 对照品适量(经折算),分别用50%甲醇水(含0.1%FA)溶解并定容至25 mL,摇匀,即得诺氟沙星储备液和诺氟沙星-d8 储备液。取诺氟沙星储备液适量,用50%甲醇水(0.1%FA)稀释,得含诺氟沙星质量浓度分别为16 000、8000、4000、2000、1000、800、400、200 ng·mL-1的系列浓度标准曲线工作液,12 800、6000、1600、600 ng·mL-1的系列浓度质控工作液及质量浓度为200 ng·mL-1的定量下限工作液。取内标诺氟沙星-d8 对照品储备液,用50%甲醇水(含0.1%FA)稀释,得质量浓度为10 μg·mL-1的内标溶液,取适量内标溶液用甲醇稀释(内标溶液∶甲醇=3∶1000),得诺氟沙星-d8 质量浓度为30 ng·mL-1的内标工作液,同时也是蛋白沉淀剂。
2.4 标准曲线样品及质控样品配制
精密吸取人空白血浆190 μL,加入系列质量浓度工作液(16 000、8000、4000、2000、1000、800、400、200 ng·mL-1)或质控工作液 (12 800、6000、1600、600 ng·mL-1) 10 μL, 涡旋混匀,得到线性范围浓度为10~800 ng·mL-1的标准曲线样品以及浓度为30(LQC)、80(MQC)、300(HMQC)、640(HQC)ng·mL-1的质控样品。
2.5 生物样品处理方法
2.5.1 空白血浆样品 精密吸取血浆样品200 µL,加入甲醇400 µL,振荡30 s,离心(18 400×g,8 min),取上清液100 μL,加稀释剂(0.2%FA 水)400 μL,振荡30 s 即得。
2.5.2 零浓度血浆样品 精密吸取空白血浆200µL,加入400 µL 内标工作液,振荡30 s,其余操作同“2.5.1”项下。
2.5.3 标准血浆样品 精密吸取标准血浆样品200µL,加入400 µL 内标工作液,振荡30 s,其余操作同“2.5.1”项下。
2.6 方法学考察
2.6.1 专属性 取空白血浆样品、定量下限(LLOQ)浓度水平的样品、只含内标的样品以及只含待测物的定量上限(ULOQ)浓度水平的样品。考察内源性物质及内标是否存在干扰。空白血浆中的内源性物质不对待测物与内标产生干扰;待测物与内标之间互相不产生干扰。结果见图1。

图1 人血浆中诺氟沙星和诺氟沙星-d8 的典型色谱图Fig 1 Typical chromatogram of norfloxacin and norfloxacin-d8 in human plasma
2.6.2 残留考察 按定量上限样品、空白样品、定量下限样品的顺序进样,记录待测物与内标的色谱峰面积值,考察残留情况。待测物进样残留小于20%且内标的进样残留小于5%。表明以本方法测试高浓度诺氟沙星人血浆样品后,在系统中的残留对于后续低浓度样品的测试无影响。
2.6.3 线性范围与定量下限考察 配制标准曲线血浆系列浓度样品,按“2.5”项下方法处理,进样分析,记录待测物与内标的色谱峰峰面积值,以诺氟沙星与内标的色谱峰峰面积比值(Y)为纵坐标,以系列标准血浆样品质量浓度(X)为横坐标,进行线性回归,得到回归方程及相关系数。
结果诺氟沙星在10~800 ng·mL-1与测定值线性关系良好,定量下限为10 ng·mL-1,回归方程为y=0.0227x-0.0037,r2=0.9995。
2.6.4 精密度与准确度 配制质量浓度为10、30、80、300、640 ng·mL-1的质控标准血浆样品各6 份,按“2.5”项下方法处理后进样测定,计算各样品浓度,计算批内和批间的精密度,结果见表1。

表1 人血浆样本中诺氟沙星的批内、批间精密度(x±s,n =6)Tab 1 Intra- and inter-batch precision of norfloxacin in human plasma (x±s,n =6)
2.6.5 提取回收率 取6 份不同来源的空白血浆190 μL,每个来源平行2 份,加入甲醇400 μL,振荡30 s,离心(18 400×g,8 min),得到6 份基质上清液,用0.2%FA 水配制含诺氟沙星低、中、高质量浓度溶液2.5、6.667、53.333(ng·mL-1)、和内标(5 ng·mL-1)。振荡30 s,混匀,进样10 μL,记录待测物诺氟沙星与内标诺氟沙星-d8 的色谱峰峰面积值。
诺氟沙星质控样品低、中、高浓度的回收率分别为82.7%,86.0%、82.7%,总体变异系数(CV)为2.30%。内标平均回收率为81.6%。
2.6.6 基质效应 取6 份不同来源的空白血浆190 μL,加入10 µL 50%甲醇水(0.1%FA)溶液,再加入甲醇400 μL,振荡30 s,离心(18 400×g,8 min),得6 份不同来源空白基质上清液备用。按空白血浆方式同样处理可得溶血基质上清、高脂基质上清。
移取空白基质上清100 µL,加入诺氟沙星低、中、高浓度溶液400 µL,振荡30 s,混匀,即得基质效应考察样本。按照空白血浆方式同样处理可得溶血血浆、高脂血浆基质效应考察样本。
取67%甲醇水溶液100 µL,加入诺氟沙星低、中、高浓度溶液400 µL,振荡30 s,混匀,即得基质效应参比样本。
人空白血浆、溶血血浆、高脂血浆均无明显基质效应。结果见表2。
表2 人血浆中诺氟沙星基质效应(x±s,n =6)Tab 2 Matrix effect of norfloxacin in human plasma ( ±s,n =6)

表2 人血浆中诺氟沙星基质效应(x±s,n =6)Tab 2 Matrix effect of norfloxacin in human plasma ( ±s,n =6)
诺氟沙星浓度/(ng·mL-1)空白血浆空白溶血血浆高脂血浆内标归一化基质因子CV/%内标归一化基质因子CV/%内标归一化基质因子CV/%30.001.07±0.021.870.99±0.055.051.00±0.066.00 80.001.04±0.032.880.97±0.022.060.98±0.033.06 640.001.11±0.032.700.98±0.033.061.01±0.021.98
2.6.7 稳定性 对样品采集、样品储存及分析过程中所涉及到的稳定性条件进行考察,结果见表3。

表3 诺氟沙星不同条件下的稳定性Tab 3 Stability test of norfloxacin under different conditons
2.6.8 稀释方法学验证 配制质量浓度为1600 ng·mL-1的诺氟沙星血浆样品,用空白血浆稀释10倍,平行处理6份,考察血浆样品的稀释可靠性。结果平均准确度偏差为-4.95%,精密度为2.1%。
2.6.9 分析批容量考察 配制标准曲线样品及低、中、高浓度质控样品,按照“2.5”项下血浆样品处理方法处理后进样分析,分析批进样数量作为分析批容量考察结果。结果表明,分析批中可以连续进样至少180 个未知样本。
2.7 测定方法在生物等效性(BE)研究中的应用
2.7.1 给药方案与血样采集 本研究经湖南省脑科医院医学伦理委员会的批准(编号为2017010)。在一项单中心、随机、开放、2×2 交叉设计的诺氟沙星BE 研究,入组8 例受试者且所有受试者均完成两周期研究,洗脱期为7 d。受试者在早餐后单次口服受试制剂100 mg(1 片)或参比制剂100 mg(1 片),240 mL 常温水送服。在给药前(0 h)以及给药后 0.25、0.5、0.75、1、1.25、1.5、2、3、4、5、6、8、10、12、24 h 进行血样采集。血样采集后置于已编号的肝素钠抗凝负压管内,轻柔颠倒混匀,4℃离心10 min(3500 r·min-1)后分离得血浆样品,-80℃冰箱保存待测。
取受试者血浆样本按“2.5.3”项下方法处理后进行测定,记录待测物质和内标的峰面积,代入回归方程,计算血药浓度。
2.7.2 数据分析 血药浓度数据采用Phoenix WinNonlin 软件(Pharsight Corporation)进行非房室模型药代动力学参数的估算分析,计算主要药代动力学参数,以全面反映药物在人体内吸收、分布、代谢和排泄的特点。
2.7.3 健康受试者的药代动力学研究 8 名受试者单剂量口服诺氟沙星受试制剂及参比制剂后血浆中诺氟沙星平均药浓度-时间曲线见图2;主要药代动力学参数见表4。AUC0~t和AUC0~∞指标均提示受试制剂与参比制剂生物等效。Cmax指标结果提示尚不能拒绝生物不等效,因本试验设计密集采血点为0.25、0.5、0.75、1、1.25、1.5 h,实际结果受试制剂tmax中位值为2.25 h,Q1~Q3 为1.5~3 h;参比制剂tmax中位值为3 h,Q1~Q3 为2~3.5 h,密集采血时间早于受试制剂和参比制剂tmax、Cmax相关数据可能非真实值。本次为预试验,样本量较小,因此Cmax相关数据需要进一步验证。
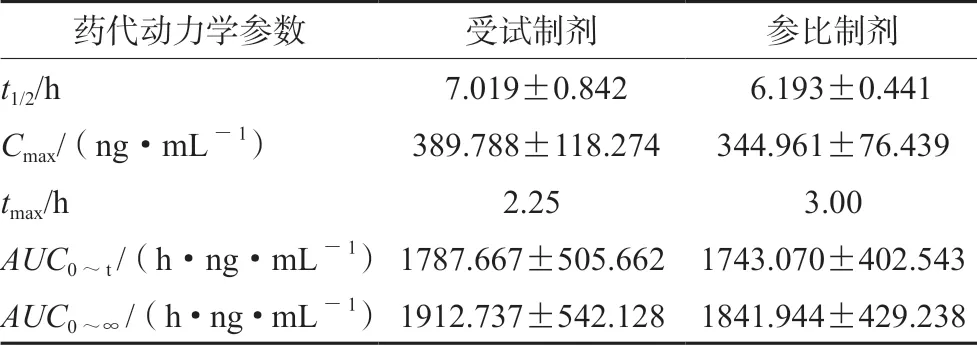
表4 8 名受试者单剂量口服诺氟沙星的主要药代动力学参数(x±s)Tab 4 Main pharmacokinetic parameters of norfloxacin in 8 healthy volunteers after a single oral dose (x±s)

图2 受试者口服受试制剂和参比制剂后血浆诺氟沙星平均血药浓度-时间曲线(n =8)Fig 2 Mean plasma concentration-time curve of norfloxacin in the test and reference preparations after the oral administration (n =8)
3 讨论
3.1 配制溶液及色谱柱的选择
本方法的关键点是根据诺氟沙星的结构特点对其色谱、质谱条件进行优化。诺氟沙星在水中溶解度较小[16],《中国药典》中也对其溶解性进行了阐述,根据这些信息,本方法选择50%甲醇水(含0.1%FA)溶液配制诺氟沙星溶液。此外,诺氟沙星属于喹啉羧酸类化合物,同时含有羧基和氨基,其logP为0.82,表明该化合物极性较大,在常规的C18柱中保留较差[17],所以在液相条件摸索过程中,选择Waters ACQUITY UPLC HSS T3(2.1 mm×50 mm,1.8 µm),该色谱柱完全耐受100%水相且对含有羧酸基团的化合物有较好的保留。
3.2 流动相的选择
本试验过程中对流动相进行了优化,水相考察了不同比例甲酸水及缓冲盐体系,通过对比,确定使用水相中含0.2%FA 最为适合。有机相分别用甲醇和乙腈进行洗脱,经过对比发现使用乙腈作为有机相时,色谱峰峰宽更窄,且响应更好。另外乙腈的洗脱能力比甲醇强,很好地避免了诺氟沙星在色谱柱中残留。
3.3 质谱条件的考察
本试验采用正离子模式,在流动相中加入适量甲酸,有助于提高离子化效率,提高响应,同时也能获得较好的色谱峰,而负离子模式则响应较低,与峰形不能同时兼顾,且文献也多采用正离子模式[14-15,18]。
3.4 内标的选择
本试验采用同位素内标,其被广泛用于化学和临床分析中的定量分析[19-20],同位素内标具有与分析物几乎相同的物理化学性质,在分析物信号波动和量的损失上的矫正作用更强,可以有效校正甚至消除基质效应的影响。为保证待测物和内标之间不存在明显干扰,本试验对同位素内标和待测物之间的通道干扰进行了考察,最终选定m/z320.11 →m/z276.13 和m/z328.17 →m/z284.20 分别作为诺氟沙星和内标的检测离子对。
3.5 分析效率的优化
本方法通过对色谱柱、流动相体系、前处理过程中稀释剂的优化,避免了残留。得益于样品前处理过程的优化,使得本方法通过简单的等度洗脱就可以实现很好的检测结果,与采用梯度洗脱的方式相比,基线更平稳,样品保留时间更稳定,也不容易出现残留,而且对仪器的要求降低,即使单液相泵也可以正常运行该方法,另外样品之间不需要设置平衡时间来让系统恢复到所需的流动相比例,可以极大地缩短进样时间,这对于样品测定来说,相同时间内可以检测更多的样品,明显提升测定效率,显著降低仪器占用成本和人工成本。本方法将分析时间缩短为1.5 min,极大地提高了分析效率,非常适合于大批量样品的分析。
3.6 小结
本次方法学验证严格按照国内外法规要求进行,验证内容完整且各项验证内容均符合验证接受标准。尤其是近年来备受关注的溶血基质效应、高脂基质效应和全血稳定性内容,本方法均进行了完整的考察,使得方法更加完善、可靠,且已用于诺氟沙星片人体BE 研究,样品测试完成后还开展了试验样品再分析(ISR)测试,ISR 合格率为100%,表明该方法可以稳定、可靠、可准确地检测人血浆样品中诺氟沙星的浓度,为诺氟沙星BE 研究提供真实、可靠的血药浓度数据。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:23:00
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
中国特种设备安全(2021年12期)2021-04-26 14:37:00
保健与生活(2019年1期)2019-01-13 13:54:39
中成药(2018年6期)2018-07-11 03:01:32
制造技术与机床(2017年9期)2017-11-27 02:14:23
中国粮油学报(2016年5期)2016-01-23 02:45:06
西南石油大学学报(自然科学版)(2015年3期)2015-04-16 05:12:08
应用化工(2014年1期)2014-08-16 13:34:08
