
LC-MS/MS法同时测定肾移植患者血浆中霉酚酸及两种代谢物浓度
2021-10-09 08:15蒋振伟杨旭萍蒋艳凌静董露露邹素兰陈荣胡楠常州市第一人民医院药学部江苏常州213003
中南药学 2021年9期
蒋振伟,杨旭萍,蒋艳,凌静,董露露,邹素兰,陈荣,胡楠(常州市第一人民医院药学部,江苏 常州 213003)
霉酚酸(mycophenolic acid,MPA)是由青霉菌发酵生成的有机弱酸,在体内通过抑制次黄嘌呤核苷酸脱氢酶的活性,阻断嘌呤核苷酸的合成,抑制T、B 淋巴细胞的增殖,从而发挥免疫抑制效应[1]。MPA 在体内主要通过尿苷二磷酸葡萄糖醛酸转移酶(uridine diphosphate glucuronyl-transferase,UGT)代谢为葡萄糖醛酸化物(MPA-glucuronide,MPAG)和酰基化的葡萄糖醛酸化物(acyl-MPAglucuronide,AcMPAG)[2],各化合物结构见图1。MPA 的药动学个体间差异较大,且浓度与疗效和不良反应有关[2],治疗药物监测具有重要意义。MPAG、AcMPAG 与MPA 游离药物浓度、肝肠循环及二次峰浓度等有密切关联[3-4],因此测定上述三者血药浓度于MPA 血药浓度监测、指导临床合理用药有实际意义。现有的关于MPA 及其代谢物血药浓度测定的方法基本是测定其中的单一化合物。同时测定MPA、MPAG、AcMPAG 有助于提高测定效率和药物检测时效。本研究建立了同时测定血浆中三者浓度的LC-MS/MS 法。
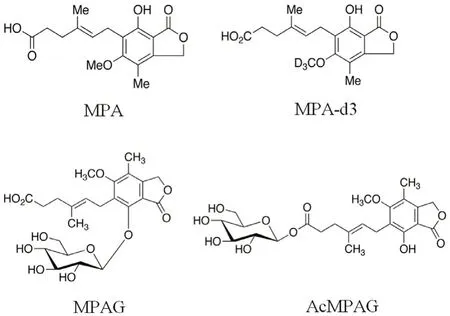
图1 MPA、MPAG、AcMPAG 及MPA-d3 的化学结构Fig 1 Chemical structure of MPA,MPAG,AcMPAG and MPA-d3
1 材料
1.1 仪器
Jasper HPLC 液相色谱仪,Triple Quad 4500MD三重四极杆串联质谱仪(AB SCIEX,美国),Vortex-Genie 2 涡旋混合器(Scientific industries,美国),5417R 低温高速离心机(Eppendorf,德国),Direct-Q 纯水仪(Millipore,美国),BT25S 电子分析天平(Sartorius,德国)。
1.2 试药
MPA(纯度:98%, 批号:3-BKG-19-1)、MPAG(纯度:96%,批号:2-FAI-5-1)、AcMPAG(纯度:95%,批号:6-PQY-99-1)(Toronto Research Chemicals 公司),内标MPA-d3(纯度:98%,批号:3-ACA-246-4,Cmass 公司)。甲醇(色谱纯,Merck公司);甲酸和乙酸铵(分析纯,Sigma-Aldrich 公司);纯水由Milli-Q 超纯水系统制得。
2 方法与结果
2.1 色谱条件
Kinetex C18(3 mm×100 mm,2.6 μm,Phenomenex,USA),流动相A:水(0.1%甲酸,10 mmol·L-1乙酸铵),流动相B:甲醇(0.1%甲酸),梯度洗脱:0~0.8 min,60%A;0.8~3.8 min,0%A;3.8~4 min,60%A;4~5 min,60%。流速:0.55 mL·min-1,柱温:40℃。
2.2 质谱条件
ESI 正离子检测模式,多反应监测(MRM)扫描。各成分选用的离子对和DP、CE 电压如下:
MPA:m/z321.0 →303.2,75 V 和15 V;MPAG:m/z514.0 →303.3,30 V 和18 V;AcMPAG:m/z514.0 →207.0,30 V 和50 V;内标MPA-d3:m/z324.1 →306.0,110 V 和15 V。
2.3 对照品储备液的配制
取MPA 5 mg、MPAG 2.5 mg、AcMPAG 0.5 mg,加入适量甲醇溶解,配制成MPA、MPAG 及AcMPAG 质量浓度分别为5、5、0.5 mg·mL-1的对照品储备液。分别取对照品储备液适量,混合配制成MPA、MPAG 及AcMPAG 质量浓度分别为0.5、2、0.4 mg·mL-1混合对照品储备液。
2.4 血浆样品处理
取血浆50 μL,加入5 μL 内标MPA-d3(10 μg·mL-1),再加入200 μL 甲醇-乙腈溶液(9∶1),涡旋震荡3 min,16 400 r·min-1离心10 min,取上清液20 μL,加入50%甲醇溶液180 μL,涡旋30 s,16 400 r·min-1离心10 min,取上清液100 μL 进样。
2.5 方法学验证
2.5.1 专属性 取6 份不同来源的人空白血浆、加入对照品储备液的人血浆样品、用药后的血浆样品按照“2.4”项下方法处理分析。结果MPA、MPAG、AcMPAG 和内标(MPA-d3)的保留时间分别为2.8、2.39、2.58 和2.79 min,血浆中内源性物质在待测物保留时间内无干扰,结果见图2。
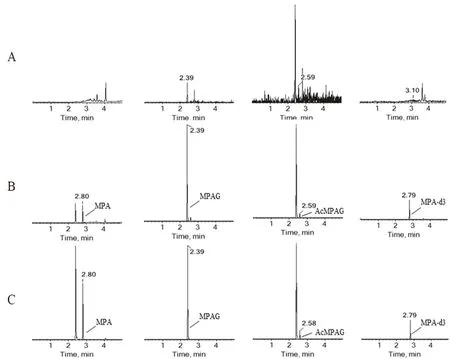
图2 血浆中MPA、MPAG、AcMPAG 和内标MPA-d3 的LC-MS/MS 色谱图Fig 2 LC-MS/MS chromatogram of MPA,MPAG,AcMPA and MPA-d3 in the plasma
2.5.2 标准曲线和定量限 精密量取混合对照品储备液适量,加甲醇稀释成含MPA(2、4、10、25、50、100、250、500 μg·mL-1),MPAG(10、20、50、125、250、500、1250、2500 μg·mL-1),AcMPAG(0.8、1.6、4、10、20、40、100、200 μg·mL-1)的混合工作溶液。取混合工作液5 μL,加入空白血浆45 μL,制备标准系列溶液。按“2.4”项下方法处理。以待测物与内标物峰面积比Y为纵坐标,待测物质量浓度X(μg·mL-1)为横坐标,以加权(W=1/χ²)最小二乘法进行线性回归运算,结果MPA 标准曲线为:Y=3.754X+0.3733(r=0.9926);MPAG标准曲线为:Y=0.4653X+0.1271(r=0.9941);AcMPAG 标准曲线为:Y=0.2891X+4.441×10-4(r=0.9942);MPA、MPAG、AcMPAG 的定量限分别为0.2、1、0.08 μg·mL-1。
2.5.3 精密度与准确度 取混合对照品储备液,配制成含MPA(5、80、400 μg·mL-1)、MPAG(25、400、2000 μg·mL-1)和AcMPAG(2、32、160 μg·mL-1)的低、中、高浓度的质控工作液。取混合质控工作液5 μL,加入空白血浆45 μL,按“2.4”项下方法处理,每个浓度取6 份,在2周内测定3 次。根据当日的标准曲线计算日内和日间精密度、准确度,结果见表1。
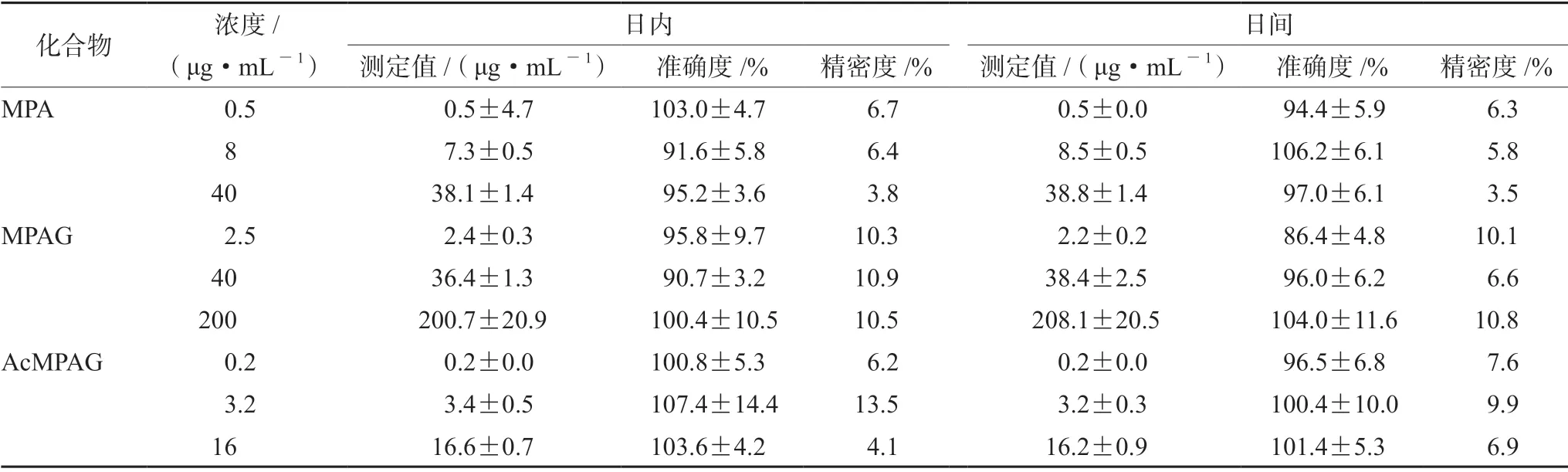
表1 MPA、MPAG、AcMPAG 的准确度及精密度(n =6)Tab 1 Accuracy and precision of MPA,MPAG and AcMPAG (n =6)
2.5.4 提取回收率和基质效应 取“2.5.3”项下3个浓度的质控QC 工作液,进样测得待测物峰面积比值A1。取空白血浆进行预处理后的上清液,加入低、中、高浓度的混合质控样品(包括内标),每个浓度6 份,测得待测物峰面积比值A2。峰面积比A1/A2可求得3 种浓度待测物的提取回收率。取不同人空白血浆6 份,加入低,中,高浓度质控样品溶液,每个浓度6 个待测样品,按“2.4”项下方法处理,测得待测物峰面积比值A3,基质效应为A2/A3,结果MPA、MPAG 和AcMPAG 的提取回收率及基质效应RSD均符合方法学要求。
2.5.5 稳定性 考察了室温稳定性,4℃稳定性,冻融稳定性及进样器稳定性。取“2.5.3”项下3个浓度的质控工作液各5 μL,每个浓度6 份,加入人空白血浆,分别在室温下放置6 h;在4℃下放置24 h;在-80℃,反复冻融3 次,按“2.4”项下方法处理后进样测定,结果MPA、MPAG 和AcMPAG 质量浓度的RSD均在±15%范围内,表明MPA 及其代谢物在本实验条件下稳定。
2.6 药动学研究
7 例肾移植患者,术后免疫抑制剂方案均采用他克莫司(FK506)+霉酚酸酯(MMF)+糖皮质激素三联用药。MMF 在肾移植术前12 h 内开始口服1000 mg bid;术中及术后1 d 静脉滴注甲基强的松龙500 mg·d-1,术后第2日或者第3日减为250 mg·d-1,后口服泼尼松片40 mg/(kg·d),逐渐减量至10~20 mg/(kg·d),他克莫司剂量为0.07~0.15 mg/(kg·d)bid。于本方案治疗1 周后,于末次给药后0、0.5、1、1.5、2、4、6、8、10、12 h 采集血浆样本测定MPA、MPAG 和AcMPAG 浓度。用WinNonlin 软件的非房室模型进行拟合求算Cmax、tmax、t1/2、AUC及平均驻留时间(MRT)等药动学参数。实验数据以mean±SD 表示,结果见表2及图3。

表2 7 例肾移植患者服用霉酚酸酯后MPA 和代谢物的药动学参数Tab 2 Pharmacokinetic parameters of MPA and metabolites after the oral administration of MMP in 7 renal transplant patients
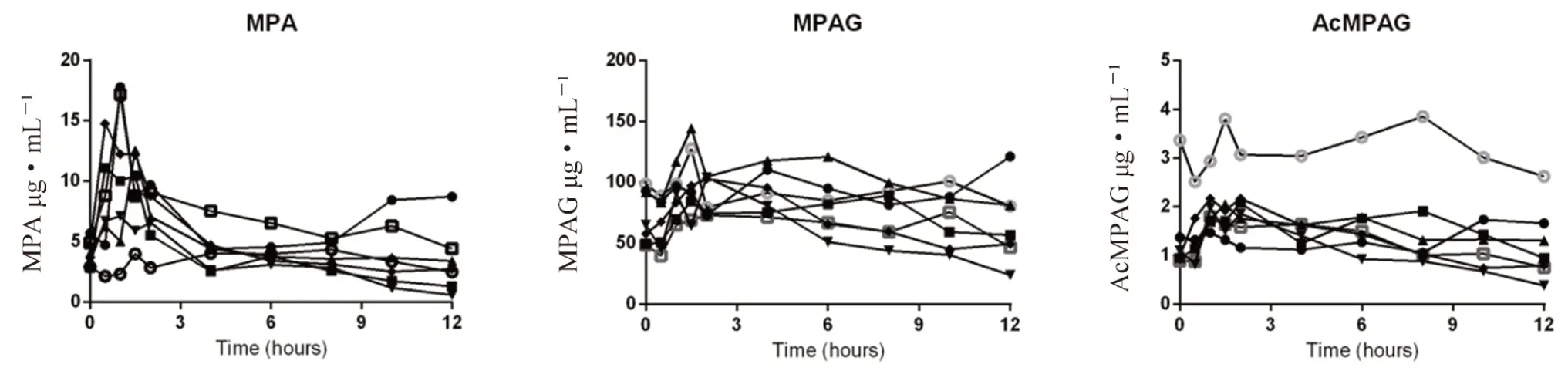
图3 肾移植患者口服霉酚酸酯后MPA、MPAG、AcMPAG 的血药浓度-时间曲线Fig 3 Mean plasma concentrations-time curve of MPA,MPAG and AcMPAG in renal transplantation patients after the oral administration of MMP
3 讨论
目前临床上应用的MPA 类药物主要有霉酚酸酯(MMF)肠溶片和霉酚酸钠肠溶片(ECMPS)[5],MMF 是无活性的前体药物,口服后经胃肠道吸收,水解生成活性代谢产物MPA。霉酚酸类药物具有非线性药动学特征,在不同患者体内药动学差异较大,需要通过血药浓度监测调整给药剂量。MPA 的血药浓度-时间曲线下面积(AUC)的理想范围在30~60 μg·h·mL-1,大于60 μg·h·mL-1时易发生胃肠道、骨髓抑制等相关不良反应,而小于30 μg·h·mL-1时发生急性排斥反应的风险增加[6]。研究表明代谢物MPAG 会影响MPA 的药动学特征,AcMPAG可能与MPA 的毒副作用有关[7-8]。MPAG 会与MPA 竞争蛋白结合影响MPA 的游离浓度。AcMPAG 可能通过抑制次黄嘌呤核苷酸脱氢酶、白细胞的增殖和诱导细胞因子的释放表现出生物活性,服用MMF 后引起的白细胞减少和胃肠道功能紊乱可能与 AcMPAG 相关[9]。因此对MPA及其代谢物的检测具有一定的临床意义。
目前国内外测定MPA 及其代谢物的方法主要有HPLC 法、LC-UV 法以及酶放大免疫法(EMIT)[10]。免疫法方便快捷,但是特异性不强,测定MPA 及其代谢物时,会与AcMPAG 发生交叉反应使MPA 浓度偏高。HPLC 法具有较高的灵敏度和特异性,是测定MPA 及其代谢物的一种标准方法,但检测过程耗时较长。本实验选择LC-MS/MS 测定MPA 及其代谢物浓度,相较于HPLC 和EMIT 法,LC-MS/MS 法不需要使用昂贵的抗体,可以缩短测定时间,样本处理方法简单,同时具有更好的特异性和灵敏度。
在方法优化过程中尝试了多种流动相,包括水-甲醇、 水-乙腈、 水(0.01%甲酸,10 mmol·L-1乙酸铵)-甲醇(0.01%甲酸)、水(0.1%甲酸,5 mmol·L-1乙酸铵)-甲醇(0.1%甲酸),最终选择流动相A:水(0.1%甲酸,10 mmol·L-1乙酸铵),流动相B:甲醇(0.1%甲酸),通过加入甲酸和乙酸铵使得MPA 及其代谢物的色谱峰形和响应值得到改善。对于洗脱方式,尝试了等度洗脱与梯度洗脱,相对等度洗脱,梯度洗脱有更好的峰形以及分离度。通过调整初始有机相比例得到了较为合适的保留时间,避免与杂质共同洗脱从而减少了基质效应的干扰。笔者查阅相关文献,发现大部分研究对于MPA、MPAG 和AcMPAG 母离子的选择都是[M-H]-离子[11-14],而在质谱条件摸索时发现MPA 的[M +H]+离子,MPAG 和AcMPAG 的[M +NH4]+离子也有很好的响应,能够满足MPA 血药浓度测定的需要。MPAG 和AcMPAG 都是霉酚酸的葡萄糖醛酸化代谢物,互为同分异构体具有相同的分子量,刘晓雪等[11]建立的肝移植患者血浆MPA及其代谢物的LC-MS/MS 检测方法中MPAG 和AcMPAG 共用同一个离子对,笔者在实验中发现了两种代谢物的不同的离子对,每个代谢物在各自的离子对下都具有较高的响应,可以更好地对MPAG 和AcMPAG 进行定性和定量。对于内标的选择,本实验选择稳定同位素内标MPA-d3,相对于吲哚美辛内标[11],MPA-d3 的理化性质与待测物最相近,表现为相似的回收率、离子化效率和色谱保留行为,并且无内源性干扰,受到基质效应的干扰更小。
猜你喜欢
云南化工(2021年10期)2021-12-21
食品安全导刊(2020年21期)2020-09-07
中国当代医药(2020年14期)2020-07-04
医学综述(2020年4期)2020-02-16
中成药(2019年12期)2020-01-04
农家科技中旬版(2019年9期)2019-10-08
山西农业科学(2019年6期)2019-06-19
中成药(2017年10期)2017-11-16
中成药(2017年5期)2017-06-13
山东工业技术(2016年13期)2016-06-29
