
结合通路信息对复杂疾病进行表型预测的SGL方法
2021-10-09 08:20徐州医科大学公共卫生学院流行病与卫生统计学系221004杨家骥余星皓黄水平
中国卫生统计 2021年4期
徐州医科大学公共卫生学院流行病与卫生统计学系(221004) 杨家骥 余星皓 曾 平 黄水平
【提 要】 目的 将整合通路信息的sparse group LASSO方法与近年来发表的表型预测方法进行比较,通过模拟各种复杂疾病可能的遗传结构,比较各方法的预测能力,期望通过TCGA数据找到高效和稳健的统计方法。方法 本研究利用SGL方法整合基因途径信息和基因表达数据,并与传统模型(LASSO、Enet、GSSLASSO)进行比较。通过乳腺癌真实基因型数据模拟表型数据:考虑不同分组(分组k=50,200,300,328)和不同遗传度对模型的影响(遗传度h2=0.3,0.5,0.8)。采用相关系数R评价几种模型的预测能力,进一步通过结直肠癌(CRC)、胰腺癌(PAAD)、乳腺癌(BRCA)三个真实数据比较各方法表型预测的准确性。结果 模拟结果表明,随着遗传度的增高,各方法的预测准确性也逐渐增高。整合通路信息的SGL方法和GSSLASSO方法比传统的LASSO和Enet方法有着更高的预测精度。而两种整合通路信息的方法中,SGL方法有着更好的预测能力和稳定性。在50,200,300分组情况下,GSSLASSO预测效果和LASSO以及Enet相近,但是在考虑通路信息的328分组下,GSSLASSO表现出了较好的预测效果。实例数据分析CRC,PAAD数据中,SGL方法具有最优的预测精度,其次是GSSLASSO,LASSO和Enet方法预测效果最差。结论 整合通路信息的预测方法预测效果明显优于一般模型,而无论是在模拟数据还是实例数据中SGL的方法具有最优的预测精度。
生物技术的飞速发展产生了大量高通量测序数据,这不仅仅为研究遗传因素与复杂疾病、特征之间的关系提供了广阔的空间,并且可以通过整合一系列组学信息,进一步促进了复杂表型遗传风险预测和评估的发展[1-6]。与传统预测模型不同,遗传预测模型研究会带来数据高维度(变量的数目p要远远大于样本量n)的问题,这就使得传统的分类和预测方法预测精度下降,计算负担加重[7]。针对高维数据,研究者通常会利用正则化的方法来提高统计模型的预测准确性和可解释性(例如LASSO、Elastic net),通过增加一个l1或者l2惩罚项对一部分模型系数进行压缩,以达到变量选择的目的。这些方法被广泛应用于大规模分子数据的疾病预测和诊断中[8-10]。
在遗传预测方面,近几年研究者们提出了许多利用分组信息进行预测的方法,绝大多数是对LASSO方法进行改进的模型选择方法,但这些正则化的方法对分组进行惩罚不可避免地会导致遗传信息的丢失。例如Yuan和Lin提出的group LASSO的方法,该方法首先将所有变量分组,然后在目标函数中惩罚每一组的l2范数,这样就可以将一整个组剔除[11]。2010年Friedman提出了一种稀疏分组LASSO(sparse group LASSO,SGL)的方法[12],这种方法对其分组和组内变量均进行正则化,以达到变量选择和模型选择双重目的[13]。另外,研究者们也发展了多种利用外部分组信息的高维数据分析方法。Tang等人在group LASSO基础上提出了一种分组的穗和板套索广义线性模型(group spike-and-slab LASSO,GSSLASSO),该方法发现在模型拟合中纳入KEGG通路信息,可以有效地提高预测的准确性[14]。本研究将SGL模型应用于连续型的高维遗传数据中,进一步整合KEGG通路信息,利用模拟研究和真实数据分析与整合分组信息的GSSLASSO模型以及不考虑分组信息的LASSO方法[15]、Enet模型[16]比较,评价其预测精度及稳定性。
方法与材料
1.方法
LASSO是把一个惩罚项加到回归系数绝对值之和上,使其满足总和小于等于一个常数的约束条件,它通过构造一个罚函数得到一个较为精简的模型,使得一些系数被压缩,使残差平方和最小化,从而能够产生某些严格等于0 的回归系数,最终得到一个解释力较强的模型。使用LASSO的原因主要有两个:一是为了提高模型的预测精度,通过将一系列回归系数设置为0,使得预测值的方差减少,因此可以提高整体的预测精度;二是为了满足模型的可解释性,通过变量选择的方法找出影响较大的变量。Enet是一种LASSO与岭回归组合后的回归分析[17-18],即将岭回归引入的l2正则项与LASSO回归引入的l1正则项组合,通过构造罚函数,使得残差平方最小,最终得到合理的模型。一方面达到了岭回归对重要特征选择的目的,另一方面又像LASSO回归那样,删除了对因变量影响较小的特征,取得了很好的效果。GSSLASSO回归是Tang等人[14]提出的一种纳入了外部注释信息的模型。该模型在系数上引入一个新的先验分布,即混合尖峰和平板双指数先验。其通过自适应的调整收缩量来提高系数估计和预测的准确性,根据不同的基因表达与表型数据,产生合适的收缩系数,去除与疾病不相关的基因,同时保留系数较大的基因,并把期望最大化步骤整合到循环坐标下降算法中,可以很好地识别重要的预测因子并从大量的候选分组中构建有效的预测模型。稀疏组LASSO(SGL)在分组LASSO的基础上增加一个l1惩罚项,既考虑到组内系数的稀疏性,也考虑到分组的稀疏性来决定选择重要的分组;类似于弹性网方法,参数α通常设置为0~1,用于在组LASSO(α=0)和LASSO(α=1)之间建立联系。
另外,在基因数据中同一个基因可能属于不同的通路,因此采用复制变量的方法对属于不同通路的基因进行复制,以达到更好的预测效果。
2.数据来源及质量控制
(1)模拟数据

(2)实例数据
数据全部来源于加利福尼亚大学基因组浏览器UCSC Xena(https://xenabrowser.net/),下载其中肿瘤基因图谱数据库(TCGA),一共包括三份癌症数据集,即乳腺癌(BRCA)、结直肠癌(CRC)、胰腺癌(PAAD)。数据包括癌症患者的临床数据和RNAseq基因表达水平数据。对于每种癌症,首先合并从原发癌组织测量的临床数据和基因表达水平的数据;然后移除了零表达值超过50%的基因并对剩余的基因表达数据进行标准化。
乳腺癌:原始数据包括1247例患者的临床数据和1218例患者的20530基因表达数据,对两份数据进行合并,删除重复的患者和男性患者,同时删除零表达值超过50%的基因,最终获得1083例患者的17675个基因表达数据。
结直肠癌:原始数据包括736例患者的临床数据和434例患者的20530基因表达数据,对两份数据进行合并,删除重复的患者,同时删除零表达值超过50%的基因,最终获得275例患者的17493个基因表达数据
胰腺癌:原始数据包括196例患者的临床数据和183例患者的20530基因表达数据,对两份数据进行合并,删除重复的患者,同时删除零表达值超过50%的基因,最终获得178例患者的18009个基因表达数据。

表1 TCGA数据集中的每种癌症的样本大小和基因数量
3.统计分析
文中所有分析均使用R 3.5.2软件,LASSO、Enet使用glmnet(version 2.0-16)软件包,通过100折交叉验证选择最优惩罚参数,Enet设置α为0.05;GSSLASSO使用BhGLM(version 1.1.0)软件包,设置s1为1,通过十折交叉验证从s0=0.01×m,m=0.1,1,2,…,9中选择最优s0;SGL使用SGL(version 1.2)软件包。基因KEGG通路注释使用clusterProfiler软件包。本研究采用100次五折交叉验证评估模型的预测精度,每次交叉验证随机抽取80%的数据集作为训练集,剩余20%作为测试集;在训练集数据中拟合预测模型,并在测试集中对连续表型进行预测,通过相关系数R评估预测性能。
结 果
1.模拟研究
图1是遗传度分别为0.3、0.5、0.8,三种模型与SGL的预测能力比较,预测性能由相关系数R衡量;每种方案重复100次。图1可以看出,与其他方法相比(LASSO、Enet、GSSLASSO),在不同的遗传度背景中SGL表现能力最佳。在分组为50的不同遗传力下,四种方法预测能力相近。而在按照KEGG分组的情况下,利用分组信息的方法明显优于传统模型,SGL预测能力最优。在50、200、300分组的情况下,LASSO、Enet和GSSLASSO预测能力相差不大。
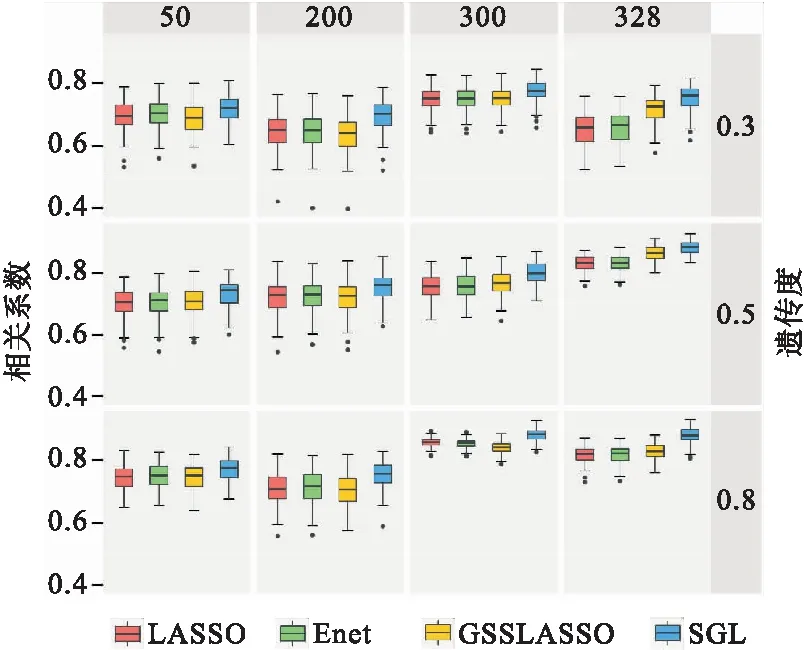
图1 三种模型与SGL的预测能力比较
2.真实数据结果
根据先前的研究,发病年龄可能是更常见于遗传起源的重要癌症指标,本研究使用乳腺癌初始病理诊断时的年龄(即发病年龄)作为表型。首先利用KEGG通路信息对基因进行分组,接着应用SGL和GSSLASSO两种方法,如果同一个基因出现在不同的通路中,就把它复制到各通路中;LASSO和Enet方法不进行分组,并删除重复的基因。
图2是使用来自TCGA数据集的三种表型对四种模型的预测性能进行比较,预测能力通过相关系数R来衡量;每种方法重复100次。从图2可以看出在两个真实数据集中,利用外部信息的SGL和GSSLASSO方法预测精度高于LASSO和Enet。在结直肠癌数据中,SGL方法预测能力最强,其次是GSSLASSO,SGL的预测精度最高,相关系数在0.25左右;其次是GSSLASSO,相关系数在0.22左右;LASSO、Enet预测能力最差,相关系数低于0.2;在胰腺癌数据中SGL方法预测能力最强,其次是GSSLASSO,SGL的相关系数在0.3左右,GSSLASSO的相关系数在0.2左右,LASSO和Enet的相关系数在0.16左右。

图2 三种表型对四种模型的预测性能进行比较
讨 论
本研究比较的四种方法均是稀疏模型,其中LASSO和Enet已被广泛应用于遗传学研究,在遗传统计学的多个方面都有较好的应用价值。目前,国内外关于遗传风险预测模型的主要研究大多数只考虑了变量的稀疏性,只是把单独的遗传位点纳入模型,没有考虑到位点之间可能存在的相关性和复杂结构。在实际情况中,经常会遇到具有分组结构的变量,如果忽视其中的分组结构,统计效能就会大打折扣。大量研究表明无论是SNP数据还是基因表达数据都具有复杂的遗传结构,与常见人类疾病相关的单个遗传变异不会直接导致疾病,而是作用于中间的分子表型或与其他遗传位点共同作用,进而引起高阶疾病特征的变化,忽略基因运作的分子网络和功能结构以及这些网络和结构变化如何导致疾病特征的变化会使得模型的预测精度不佳[19-21]。
SGL和GSSLASSO方法作为分组稀疏模型,它们考虑到了组间的稀疏性和组内的稀疏性。从模拟结果和真实数据结果我们可以看出,对组内基因进行惩罚可以提高预测精度。一般来说,模型的预测能力取决于数据的真实结构,模型假设与数据结构的吻合度越高,预测准确性越高。从模拟数据的分析结果可以看到各模型在不同遗传度和不同分组设置下预测能力的表现。本研究共有12种模拟方案,在9种方案中,SGL的预测能力要明显优于其他方法,在其余方案中,四种模型预测能力相近。当分组数设置为200组时,SGL模型明显优于其他方法,当分组数设置为50/300时,几种模型的预测精度接近,SGL模型要略好;当分组设置为328组,并且数据通过基因进行分组时,SGL模型有较强的预测能力和稳定性。真实数据的结果也可以看出,三种整合外部注释信息的SGL和GSSLASSO方法要优于LASSO和Enet方法。
本研究从模拟和实际出发,探索整合外部注释信息能否提高对连续型表型的预测准确性,通过对比分析,整合外部注释信息可以显著提高对遗传表型的预测,并且SGL方法有较高的预测精度。
猜你喜欢
区域治理(2022年40期)2022-11-27
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
小学生学习指导(低年级)(2019年3期)2019-04-22
小学生学习指导(低年级)(2018年9期)2018-09-26
现代园艺(2017年21期)2018-01-03
小学生导刊(低年级)(2017年1期)2017-06-12
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
