
图卷积神经网络在组学数据分类预测中的应用*
2021-10-09 08:16张刘超荣志炜赵薇薇
中国卫生统计 2021年4期
张刘超 荣志炜 赵薇薇 李 康△
【提 要】 目的 探讨图卷积神经网络(graph convolutional neural network,GCNN)利用PPI网络对组学数据的分类预测效能。方法 通过模拟实验和实例研究,对GCNN、随机森林、支持向量机和多层感知机共四种方法的分类效果进行比较。结果 模拟实验结果显示,即便在样本量和网络中节点数量较少时,GCNN的分类效能也明显优于其他三种方法,并且随着节点数量的增加而不断提高。实例研究表明,利用STRING网络,GCNN的分类效能最优。结论 GCNN在组学数据的研究中极具潜力,值得进一步研究。
肿瘤的发生发展是基因突变、表观遗传学改变,以及环境因素等共同作用的结果[1]。图卷积神经网络(GCNN)[2]是一种适用于图(网络)结构数据的深度学习算法,即利用已知的PPI(protein-protein interaction)网络提供的基因相互作用关系,可以更有效的提取肿瘤样本的数据特征,再使用深度学习建立判别模型,有利于提高模型的判别能力,实现更好的分类预测效能。本研究通过模拟实验探究GCNN方法的分类预测效能,并与多层感知机(MLP)、随机森林(RF)和支持向量机(SVM)三种方法进行比较,最后给出应用实例。
方法和原理
1.基本原理
图结构数据[3]由网络图和节点特征值组成,网络图是指根据特定的生物学关系,如基因调控或蛋白互作关系等构成的网络图形,可以用G=(V,E,A)表示,其中V表示节点,E为连接边,A为加权邻接矩阵,每个节点有其对应的特征值。在PPI网络中,节点代表蛋白质或其对应的基因,连接边表示基因间的相互作用关系,特征值即为基因或蛋白质的表达值。图卷积神经网络算法的思想[2]:对网络各节点与其直接相连或间接相连的节点表达值多次做加权平均,由此得到的节点表达值则更加稳定;其权重可以利用拉普拉斯矩阵L,即计算各节点梯度的散度,其计算周围点与中心点的梯度差,得到的是对该点进行微小扰动后可能获得的总变化,以此作为卷积核的函数实现上述计算;最后以卷积的结果作为输入,利用全连接神经网络实现判别和分类(流程如图1)。

图1 图卷积神经网络(GCNN)流程示意图
2.具体计算过程
给定无向网络图G=(V,E,A),其中V={X1,
X2,…,Xm}表示图中节点的集合,m为图中节点的个数;E={(Xi,Xj)},(Xi,Xj)表示节点Xi与Xj之间的连接边(1≤i,j≤m);A为加权邻接矩阵,即
(1)
式中0≤aij≤1表示连接边(Xi,Xj)上的权重,aii=1。同时定义对角阵
(2)
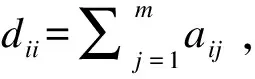
Defferrard[5]提出使用切比雪夫多项式计算图卷积操作的卷积核,若取最高为K阶的邻接点做卷积,K=0,1,2,…,m-1,则相应的卷积核为一个K维向量:
(3)

(4)
其中,n表示样本个数,m表示基因个数。则图卷积操作为
(5)

在上述卷积运算基础上,以末次卷积运算的结果X(C)作为输入,由后端的全连接神经网络进行判别和分类。
模拟实验
1.GCNN的超参数设置
图卷积神经网络(GCNN)包含两个卷积层和两个全连接层;在模拟实验和实例研究1中,每个卷积层设置32个卷积核,且K=8;后端的两个全连接隐藏层神经元个数分别为1024和516。在实例研究2中,两个卷积层分别设置32和64个卷积核,K=14;后端的两个全连接隐藏层神经元个数分别为1024和512。学习率为0.001,使用ReLU函数进行非线性转换,ReLU(x)=max(0,x)。计算样本标签真实值与预测值的交叉熵损失,使用反向传播算法对权值进行更新。
2.模拟数据
模拟实验1:两组多变量数据均值向量相同,协方差阵不同时四种方法的比较。设置变量数依次为100、250、500、750、1000,各变量的边际分布服从正态分布N(0,1),并且平均与其他10个变量相关。为了描述所有变化情况,将取自正态分布N(0.1,0.12)的数值随机与1,-1或0相乘得到不同两组的相关系数。训练集的样本量为200∶200,测试集的样本量为2000∶2000。重复实验1000次。
模拟实验2:两组多变量数据均值向量不同,同时协方差阵不同时四种方法的比较。固定变量数为500,生成一组数据各变量边际分布服从N(0,1),另一组数据各变量边际分布服从N(μ,1),均值μ分别为0、0.05、0.1、0.15和0.25。其他条件与模拟实验1相同。
3.模拟实验结果
模拟结果显示,即使在组间均值没有差异的情况下,当网络中节点数量较少时,GCNN具有较好的分类预测能力,并且随着节点数量的增加,GCNN分类预测的AUC值和准确率不断增加,并趋近于1,明显优于其他三种方法(见图2)。当固定网络图中节点数量为500时,随着两组数据间均值向量的差异不断变大,各种方法的分类预测效能相应提高,但是GCNN的分类预测效能仍优于其他方法(图3)。上述模拟实验表明,GCNN的优势在于通过利用网络表示的变量之间的相关关系,可以很好地学习到不同样本之间的特征,实现较好的分类预测效能,尤其适用于样本均值向量差异较小的情况。

图2 不同网络节点数下四种方法的比较

图3 不同组间差异下四种方法的比较
实例应用
实例研究1:为了进一步验证GCNN对真实数据的分类效能,选用TCGA数据库中黑色素瘤(SKCM)的mRNA表达数据,探究GCNN对黑色素瘤原发癌和癌转移的分类预测效能。使用OncoGenomic Landscapes数据库[7]给出的黑色素瘤相关基因进行变量筛选,选取STRING数据库[8]中相应的PPI网络。最终,本研究纳入472例黑色素瘤患者,其中368名癌转移患者,104名原发癌患者,PPI网络中含有272个节点,17687条边。使用十折交叉验证测试各个模型的分类预测效能。在GCNN和MLP的模型训练过程中,从训练集中随机选取10%的样本作为验证集,辅助模型训练。
实例研究2:选用TCGA数据库中黑色素瘤(SKCM)的蛋白质组数据和STRING数据库中的PPI网络,进一步探究GCNN在蛋白质组学上的学习效能。在剔除存在缺失的蛋白质后,本研究共纳入258名癌转移患者,96名原发癌患者,PPI网络中含有67个节点,1926条边。使用十折交叉验证测试各个模型的分类预测效能。在GCNN和MLP的模型训练过程中,从训练集中随机选取10%的样本作为验证集,辅助模型训练。
实例研究结果:由表1和表2所列结果可知,无论是转录组数据还是蛋白质组数据,GCNN对黑色素瘤癌转移预测的AUC均值为87.46%和83.30%,均高于其余三种方法,并且分类预测效能较稳定。

表1 基于mRNA表达数据的黑色素瘤转移分类预测结果(%)

表2 基于蛋白质组数据的黑色素瘤转移分类预测结果(%)
讨 论
与传统机器学习方法通过样本数据的数字特征进行分类预测相比,GCNN在研究基因或蛋白质表达量差异的同时,利用PPI网络所提供的基因间相互作用关系,通过其强大的非线性拟合能力,将样本数字特征和生物学知识进行有机结合,实现更优的分类预测效能,在组学数据的研究中极具潜力。
由模拟实验1的结果可知,当两组的均值相同时,传统机器学习方法在只考虑样本数据的数字特征时,很难区分两组样本。但是,GCNN通过利用网络结构提供的变量间的相互作用关系,仍可以学习到不同组别样本的特征,具有较好的预测效能。随着变量个数的增多,变量间的关系更为复杂,但GCNN的预测效能却在不断提高,展现出GCNN强大的拟合能力。模拟实验2的结果显示,当两组样本的均值差异较大时,虽然传统方法仅依据样本的数字特征便具有不错的区分能力,但是GCNN的分类预测效能仍然略优于传统机器学习方法,这与实例研究的结果相吻合,在黑色素瘤原发癌和癌转移患者的基因表达情况差异较大的情况下,可以看出GCNN的预测效能仍略优于RF和SVM等方法。
本研究尚存在一些不足之处,首先本研究使用OncoGenomicLandscapes数据库中的黑色素瘤相关基因进行变量筛选,尚不能使用GCNN完成变量筛选。其次,本研究所用的PPI网络并不能完全表征基因间的全部相互作用关系,可能会对GCNN造成一定程度的干扰。我们将在后续的研究中进一步解决上述问题。
猜你喜欢
北京航空航天大学学报(2021年9期)2021-11-02
石油地质与工程(2019年3期)2019-09-10
电子制作(2019年11期)2019-07-04
北京航空航天大学学报(2018年1期)2018-04-20
材料科学与工程学报(2016年2期)2017-01-15
西南国防医药(2016年6期)2016-12-01
癌变·畸变·突变(2016年5期)2016-08-22
癌症进展(2016年10期)2016-03-20
实用手外科杂志(2015年4期)2015-08-27
中国海上油气(2015年3期)2015-07-01
