
苯并呋喃酮的高效液相色谱分析
2021-10-09 03:10李春艳庞怀林
世界农药 2021年9期
王 怡,李春艳,庞怀林
(1.上海泰禾化工有限公司,上海 201615;2.南通泰禾化工股份有限公司,南通 226407)
苯并呋喃酮,又名为2-苯并呋喃酮、苯并呋喃-2(3H)-酮、2-香豆冉酮,其结构式见图1。苯并呋喃酮是许多有机合成原料、天然产物分子和生物药物中间体的重要结构单元,可广泛用于药物、食品和材料等领域[1-3],而且还是农业上高效广谱杀菌剂嘧菌酯合成工艺的重要中间体[4]。

图1 苯并呋喃酮的结构式
目前对苯并呋喃酮的检测方法较少,仅见张文娟测定苯并呋喃酮原药的相关研究[5],此方法简练高效,但因其峰保留时间短,一般能满足原药的分析,但无法适用需分离衍生物、杂质的苯并呋喃酮试样。为广泛地适用多规格产料需求,建立一种兼顾快速分析与杂质有效分离的苯并呋喃酮分析方法十分必要。考虑到嘧菌酯工艺生产过程中与邻羟基苯乙酸、对甲苯磺酸等原料的分离,一般选择乙腈与水混合溶剂作为流动相体系,或在有机相中加入磷酸或甲酸等[6]以调节pH和改善峰型,提高后续合成中与嘧菌酯的分离检测。基于上述特点,本文采用高效液相色谱法,通过对不同流动相组合及分析条件的探索和验证,确定了苯并呋喃酮测定中流动相优先选择乙腈+磷酸水混合溶液,并加入了一定比例的甲醇以改善拖尾现象,提高与杂质的分离度和选择性,建立了苯并呋喃酮的高效液相色谱测定方法。该方法具有分离效果好、操作简单快速的特点,适用于苯并呋喃酮试样的定量分析。
1 材料与方法
1.1 仪器与试剂
LC-20AD型高效液相色谱仪配SPD-M20A二极管阵列检测器,及色谱工作站Labsolution (日本岛津公司);ML204型电子天平(瑞士梅特勒托利多集团);2200T型超声波清洗仪(上海安谱科学仪器有限公司);Direct 8型纯水超纯水一体机(德国默克集团);滤膜:0.45 μm (有机相,上海安谱实验科技股份有限公司)。
乙腈,色谱纯(上海安谱实验科技股份有限公司);甲醇,色谱纯(上海安谱实验科技股份有限公司);磷酸,分析纯(国药集团);苯并呋喃酮标样,已知质量分数99.0% (南通泰禾化工有限公司提供,上海晓明检测技术服务有限公司定值);苯并呋喃酮试样样品(南通泰禾化工股份有限公司)。
1.2 色谱条件
检测器为二极管阵列检测器,色谱柱为岛津WondaSil-WR C18色谱柱(250 mm×4.6 mm i.d.不锈钢柱,内装5 μm填充物);流动相为乙腈-甲醇-0.1%磷酸水溶液,经滤膜过滤和脱气,采用梯度洗脱(表1),流速为1.0 mL/min;检测波长为268 nm;柱温为(35±2) ℃;进样体积为10 μL。在上述色谱操作条件下,苯并呋喃酮的保留时间约为10.1 min。苯并呋喃酮标样和试样的高效液相色谱图见图2、图3。

表1 梯度洗脱时间程序设置
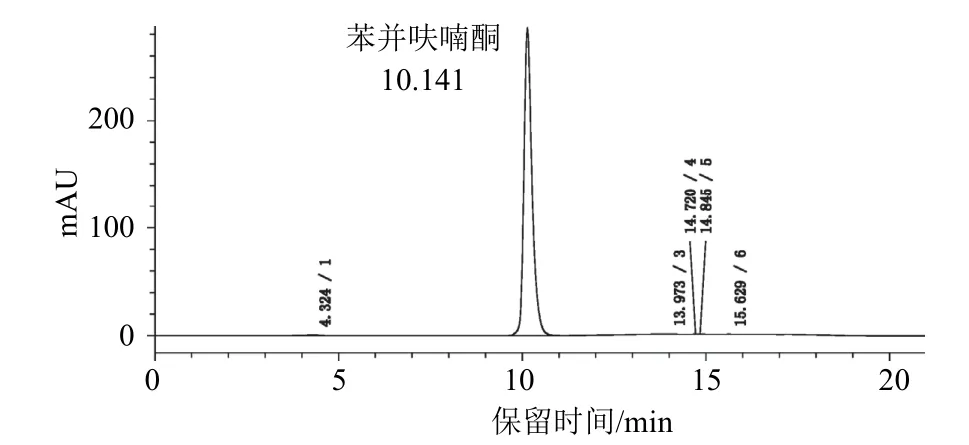
图2 苯并呋喃酮标样的高效液相色谱图(1 000 μg/mL)
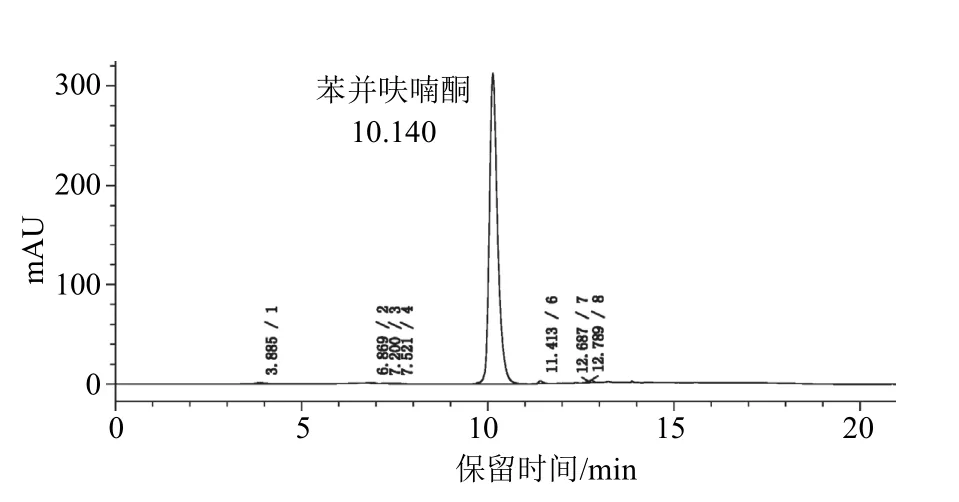
图3 苯并呋喃酮试样的高效液相色谱图 (1 000 μg/mL)
1.3 溶液的配制
1.3.1 标样溶液的配制
称取约0.06 g (精确至0.000 1 g)苯并呋喃酮标样置于50 mL容量瓶中,用流动相溶解并超声,待溶液冷却至室温后用流动相稀释至刻度,摇匀备用。
1.3.2 试样溶液的配制
称取约0.08 g (精确至0.000 1 g)苯并呋喃酮试样置于50 mL容量瓶中,用流动相溶解并超声,待溶液冷却至室温后用流动相稀释至刻度,摇匀备用。
1.4 测定
在上述操作条件下,待仪器基线稳定后,连续注入数针标样溶液,待相邻2针的峰面积相对变化小于1.0%,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进样测定。
1.5 计算
将测得的2针试样溶液以及试样前后2针标样溶液中苯并呋喃酮峰面积分别进行平均。试样中苯并呋喃酮的质量分数X(%)按式(1)计算:

式中:X为试样中苯并呋喃酮的质量分数(%);A2为试样溶液中苯并呋喃酮峰面积的平均值;m1为标样的质量的数值(g);P为标样中苯并呋喃酮的质量分数(%);为标样溶液中苯并呋喃酮峰面积的平均值;m2为试样的质量的数值(g)。
2 结果和分析
2.1 流动相的选择
为实现快速分离检测,对乙腈、甲醇、纯水、磷酸水溶液等多种有机相+水相组合,在C18色谱柱上进行比较选择,经试验比较确定流动相为乙腈和0.1%磷酸水溶液体系为主,加入甲醇的情况下,峰型不对称、拖尾的情况有较大改善;与加入5%、8%、12%的甲醇相比,在加入10%甲醇时,与杂质分离度高、保留时间短,可较大程度提高检测效率。
2.2 分析方法特异性
本试验采用HPLC-DAD峰纯度分析法来鉴别苯并呋喃酮。苯并呋喃酮标样的最小峰纯度相似度为1.00,最小峰纯度阈值为1.00,最小峰纯度指数为0,说明标样色谱峰中不含有杂质峰;苯并呋喃酮试样中的苯并呋喃酮HPLC-DAD峰纯度中最小峰纯度相似度为0.999 9,最小峰纯度阈值为0.999 9,最小峰纯度指数为4,说明样品色谱峰中不含有杂质峰。标样与试样的色谱峰保留时间差在1.0%以内,如图2和图3所示。标样和试样的峰纯度图如图4、图5所示。

图4 苯并呋喃酮标样中的目标峰纯度

图5 苯并呋喃酮试样中的目标峰纯度
2.3 分析方法的线性相关性的测定
以苯并呋喃酮质量浓度(x)为横坐标,峰面积(Y)为纵坐标,绘制苯并呋喃酮的标准曲线,结果见图6。可见,当苯并呋喃酮质量浓度为231.66~2 084.94 μg/mL,与其相应的苯并呋喃酮峰面积之间呈现出良好的线性关系,其线性回归方程为Y=3 828x+43 666,相关系数R2=0.999 9。
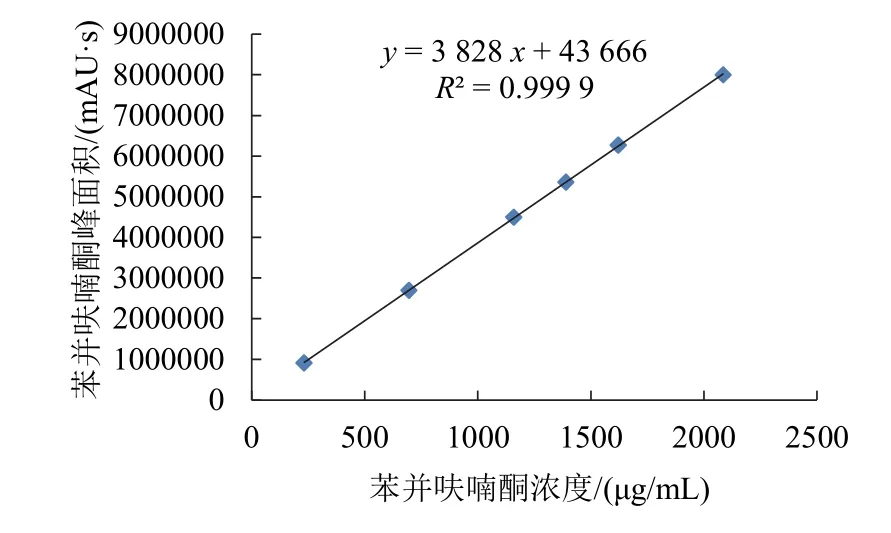
图6 苯并呋喃酮的质量浓度与其相应峰面积的线性关系图
2.4 分析方法的精密度试验
对同一试样准确连续称取6个试样,在上述的色谱操作条件下进行分析,测得苯并呋喃酮的质量分数,试验结果详见表2。可见,苯并呋喃酮的标准偏差和变异系数分别为0.09和0.13%,说明此分析方法精密度良好,能满足日常的定量分析。
2.5 分析方法的准确度试验
称取约0.04 g (精确至0.000 1 g)苯并呋喃酮试样置于50 mL容量瓶中,分别加入不同质量的苯并呋喃酮标样(精确至0.000 1 g)于同一50 mL容量瓶中,用流动相溶解并超声,待溶液冷却至室温后用流动相稀释至刻度,摇匀后,按1.2节色谱操作条件进行测定,结果见表3。可见,苯并呋喃酮的回收率为99.79%~100.22%,平均回收率为100.02%,说明该方法准确度良好,能满足日常的定量分析。
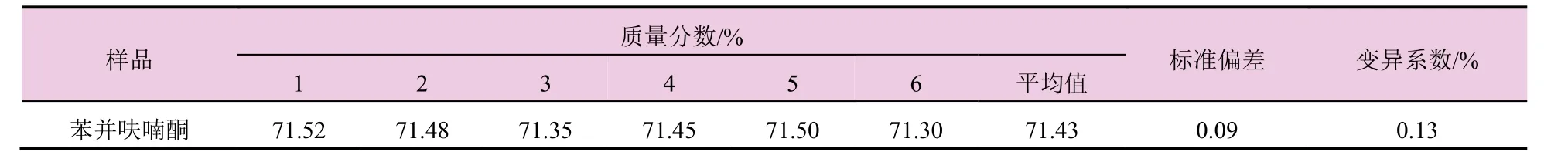
表2 苯并呋喃酮试样的精密度试验

表3 苯并呋喃酮的准确度试验
2.6 非分析物干扰试验
在评价时,需要包含非分析物的干扰,防止有效成分中存在其他干扰物导致分析方法出现误差。以空白溶剂为不含有效成分的空白样品,其高效液相色谱图如图7所示,经与图2、图3对比发现空白溶剂中不含有效成分的干扰物质。

图7 苯并呋喃酮溶剂空白的高效液相色谱图
3 结 论
本文采用高效液相色谱法建立了快速测定苯并呋喃酮试样中苯并呋喃酮的定量定性方法。该方法采用C18不锈钢色谱柱和二极管阵列检测器,乙腈、甲醇和0.1%磷酸水为流动相,测得苯并呋喃酮的精密度和准确度高、线性关系良好,具有操作简便、检测过程稳定、结果准确、分离效果好等特点,可用于苯并呋喃酮生产过程中的质量控制和分析检测。
猜你喜欢
农药科学与管理(2022年6期)2022-08-03
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
粉末冶金技术(2021年3期)2021-07-28
中国食品(2020年16期)2020-08-31
——第二部分:原棉短纤维率标样的验证试验分析
中国纤检(2020年7期)2020-07-22
食品界(2018年8期)2018-09-03
中学生数理化·高二版(2016年6期)2016-05-14
同位素(2014年2期)2014-04-16
中国信息化·学术版(2013年3期)2013-06-25
