
布洛芬缓释胶囊在中国健康受试者中生物等效性研究*
2021-10-09 04:21陈新民刘杰谢斌杨沛欣杨琼
医药导报 2021年10期
陈新民,刘杰,谢斌,杨沛欣,杨琼
(珠海润都制药股份有限公司,珠海 519041)
布洛芬为非甾体类抗炎药,其抗炎、镇痛、解热效果良好,临床主要用于风湿及类风湿性疾病,以及肌肉痛、神经痛、关节痛等多种中慢性钝痛。口服布洛芬吸收迅速,但消除半衰期较短,维持治疗常需频繁给药,长期服用可引起胃肠道刺激反应[1-2],如腹胀、消化不良等,此外还会加重肾脏损伤。缓释胶囊可有效避免患者日间频繁给药的困扰,仿制药价格往往低于原研药品,因此,对于众多的疼痛病患者减轻治疗负担具有一定意义。珠海润都制药股份有限公司生产的布洛芬缓释胶囊为化学药品仿制药口服固体制剂,已被列入首批2018年底前须完成仿制药一致性评价的289个品种目录中,需要对其进行质量和疗效与原研药(参比制剂)一致性评价工作,开展仿制药质量和疗效与原研药一致性评价工作,对提升我国制药行业整体水平,促进医药经济结构调整和产业升级,提高医药产业国际竞争能力,满足公众用药需求,都具有十分重要的意义。按照《普通口服固体制剂参比制剂选择和确定指导原则》选取中美天津史克制药有限公司的布洛芬缓释胶囊(商品名:芬必得®)为参比制剂,拟通过两项试验(空腹和餐后两种给药条件下)评估珠海润都制药股份有限公司生产的布洛芬缓释胶囊与原研制剂的生物等效性。
1 试药与仪器
1.1试药 受试制剂:布洛芬缓释胶囊,规格:300 mg,批号:3091604015,珠海润都制药股份有限公司生产;参比制剂:布洛芬缓释胶囊(商品名:芬必得®),规格:300 mg,批号:16080190,中美天津史克制药有限公司生产。布洛芬对照品,纯度:98%,批号:10-XJZ-87-1,来源Toronto Research Chemicals;布洛芬-d3(内标),纯度:99.4%,批号:1503-016A1,来源:TLC Pharmachem。乙腈,色谱级,默克公司;甲醇,色谱级,默克公司;乙酸铵,色谱纯Sigma-Aldrich。
1.2仪器 BT125D电子天平[赛多利斯科学仪器(北京)有限公司,感量:0.01 mg];XP6电子天平[梅特勒-托利多仪器(上海)有限公司,感量:0.001 mg];质谱仪API 4000(AB Sciex公司);LC-20AD 高效液相色谱仪,Shimadzu公司;Analyst®数据采集软件(版本1.6.3);离心机5810R Eppendorf;药动学分析软件WinNonlin(Pharsight Corporation,Version 6.3),描述性统计分析采用SAS 软件(9.4版本)。
2 方法与结果
2.1研究对象 本研究共入组中国健康受试者48例,空腹餐后各24例。空腹试验入组24例,男、女各12例;年龄19~44岁;体质量45.0~75.0 kg;身高145.5~184.0 cm;体质量指数19.5~24.9 kg·(m2)-1。餐后试验入组24例,其中男16例,女8例;年龄18~54岁;体质量50.0~85.5 kg;身高149.5~181.5 cm;体质量指数19.7~26.0 kg·(m2)-1。入组的受试者必须充分理解,并自愿签署本研究的知情同意书;从签署知情同意书开始至试验结束后6个月内,受试者或其伴侣必须愿意使用医学上可接受的避孕方法,且从签署知情同意书开始至试验结束受试者不能使用避孕药物;受试者能够与研究者作良好的沟通,并能够依照方案规定完成研究。试验开始前受试者均签署书面知情同意书。
2.2给药方案与血样采集 本研究是一项空腹与餐后给药,单中心、单剂量、两制剂、两周期、两序列、随机、开放、自身交叉的平均生物等效试验,清洗期为7 d。
受试者在每周期给药前1天(第1天和第7天)18:30前入住I期临床试验病房,统一生活管理,禁止统一饮食以外的任何食物和饮料。受试者于给药前1天晚餐后,禁食不禁水。在每次入住临床试验中心前,均询问受试者的健康状况和用药情况以及饮食、吸烟、饮酒、毒品等,并进行生命体征测量、烟检、酒精检测、药物滥用筛查,女性受试者查血确认是否妊娠。餐后给药要求受试者进食高脂肪、高热量餐,30 min内进食完毕。
于试验第1天、第8天约8:30,由经过培训的研究人员按随机表发放受试制剂或参比制剂,受试者空腹用药(至少空腹10 h)/餐后试验要求受试者进食高脂肪、高热量餐,30 min内进食完毕,温水240 mL送服。给药前后1 h内禁止饮水(除服药时给予的240 mL水),给药后保持上身直立状态2 h,给药后4,10 h,分别进食午餐和晚餐。
空腹和餐后试验分别在给药前0 h(给药前60 min内)和给药后0.5,1,2,3,3.5,4,4.5,5,5.5,6,7,8,10,12,14,24 h,采集前臂静脉血4 mL,置于含有肝素锂抗凝剂的采血管中,离心(1500×g,2~8 ℃)10 min,分装血浆样本,并在-70 ℃储存待测。从血样采集至血浆冻存的时间不超过2 h。服药后2 h内采血点均在预定时间±3 min内采集;服药后3~12 h采血点均在预定时间±5 min内采集;服药后14~24 h采血点均在预定时间±10 min内采集。于每周期入住时、给药前0 h(给药前60 min内)及给药后1,3,5,12,24 h以及任何研究医生认为有需要的时候,测量受试者的脉搏、血压、呼吸频率、腋下体温。
2.3方法学考察与评价
2.3.1色谱条件 色谱柱:CAPCELL MG C18(50 mm×2.0 mm,5 μm),流动相A:1.00 mmol·L-1乙酸铵溶液,流动相B:1.00 mmol·L-1乙酸铵乙腈/水(95/5)梯度洗脱,进样器温度6 ℃,柱温40 ℃,进样量5.0 μL。
2.3.2质谱条件 离子源:ESI;离子化方式:负离子模式;扫描方式:MRM;MRM离子对:布洛芬(m/z)为205.4/161.3,布洛芬-d3(m/z)为208.5/164.4。见图1。

A.布洛芬;B.布洛芬-d3。
2.3.3布洛芬血浆样品处理 精密转移血浆样品50.0 μL至96孔板中,加入300 ng·mL-1内标工作溶液50.0 μL,乙腈溶液400 μL,在摇板机上摇约5 min使充分混匀。在离心机中离心10 min(温度4 ℃,3220×g);转移上清液100 μL到一块新的96孔板中,加入50%甲醇溶液100 μL,在摇板机上摇约5 min使充分混匀。在离心机中离心5 min(温度4 ℃,3220×g),取5.0 μL进样,进行LC-MS/MS分析,并记录色谱图。
2.3.4系统适用性 系统适用性评价样品为6个系统适用性定量下限样品,在每个分析批开始进样前均进行系统适用性评价测试,以确认该仪器状态。结果显示分析物保留时间的RSD≤0.8%,内标保留时间的RSD≤0.9%,分析物与内标峰面积比的RSD≤6.5%,表明系统适用性良好。
2.3.5选择性 不同来源的空白基质对分析物均无干扰;内标对分析物、分析物对内标的检测无干扰,均符合接受标准。见图2。

A.空白基质;B.标准样品;C.受试者样品。
2.3.6标准曲线与定量下限 在1.5 mL聚丙烯离心管中,用移液器加入适量空白人正常血浆。加入相应的源溶液,涡旋混匀(约30 s),配制成相应的校正标样(浓度为50.0,100,400,2000,8000,16 000,20 000,25 000 ng·mL-1)。按样本处理操作,以分析物(布洛芬)与内标(布洛芬-d3)的色谱峰面积比对分析物浓度(权重1/χ2)进行线性回归,得回归方程Y=0.002 452X+0.017 31,线性拟合度R2为0.995 1,线性范围为50.0~25 000 ng·mL-1,定量下限50.0 ng·mL-1。
2.3.7残留效应 在最高浓度的校正标样后跟随空白基质样品来评价残留效应。在分析物保留时间处干扰峰的响应均低于定量下限中分析物响应的20%,内标保留时间处干扰峰的响应均低于零浓度样品中内标响应的5%。结果表明,最高浓度后面的空白基质样品中布洛芬≤18.4%,布洛芬-d3≤1.0%,不影响定量。
2.3.8基质效应 在96孔板中加入空白单人血浆50.0 μL,按照血浆样品处理过程处理,提取上清液后加入低、中、高浓度中间溶液(N-LQC,N-MQC,N-HQC)100 μL。中间溶液配制:精确量取LQC(150 ng·mL-1),MQC(12 500 ng·mL-1),HQC(22 500 ng·mL-1)工作液(精密称取布洛芬10.40 mg,经质量校正后,溶于100%甲醇1020 μL,得到浓度为10.0 mg·mL-1的储备液;分别量取适量储备液,用50%甲醇稀释得到相应浓度的工作液)20.0 μL和内标工作液混合400 μL,使用50%甲醇溶液3580 μL,分别稀释获得N-LQC,N-MQC,N-HQC中间溶液。使用生物基质相面积和溶剂相面积比较得到待测物和内标面积比,获得归一化基质效应因子。实验结果显示,布洛芬高、中、低3个浓度水平平均归一化基质效应为1.00,1.01,1.00,表明无明显基质效应。
2.3.9布洛芬精密度与准确度 配制定量下限、低、中和高4个浓度的质控样品(分别为50.0,150,12 500,22 500 ng·mL-1),按血浆样本处理操作,在3个分析批中,各浓度质控样品均配制6个平行样,评价本方法的精密度与准确度,结果见表1。

表1 布洛芬在人血浆中的精密度与准确度
2.3.10布洛芬的回收率 在96孔板中加入空白混合人血浆50.0 μL,按照样品处理过程处理,提取上清液后,加入低、中、高中间溶液(N-LQC,N-MQC,N-HQC)100 μL和内标工作液400 μL混合,使用50%甲醇溶液3580 μL,分别稀释获得N-LQC,N-MQC,N-HQC中间溶液。使用提取后样品峰面积和空白血浆提取的上清与中间溶液混合所得样品的峰面积分别获得待测物的回收率。结果显示,低、中、高浓度的回收率分别为84.0%,84.2%,87.5%,表明回收率良好。
2.3.11稳定性 分别考察分析物储备液在室温条件下放置23.5 h稳定性(RSD≤2.8);分析物工作液在室温条件下放置23.5 h稳定性(RSD≤4.3);全血中分析物在室温(RSD≤4.3%)条件下放置2 h稳定性;血浆中分析物在室温下放置26 h稳定性(RSD≤1.6%);处理过样品在自动进样器(6 ℃)下存储26.7 h稳定性(RSD≤4.2%);血浆中分析物经过5次冷冻和融化循环后稳定性(RSD≤6.2%);血浆中分析物储存在-20 ℃条件下107 d稳定性(RSD≤6.6%),在-80 ℃条件下107 d稳定性(RSD≤5.2%)。结果表明布洛芬的血浆样品稳定性良好。
2.4生物样本再分析ISR 本研究分析的样品总数为1615个,其中177个样品被选做已检测样品再分析(ISR),176样品符合接受标准,通过率为99.4%。
2.5血药浓度-时间曲线 空腹和餐后条件下各有24例健康受试者单剂量口服受试制剂和参比制剂,布洛芬血药浓度-时间曲线见图3。
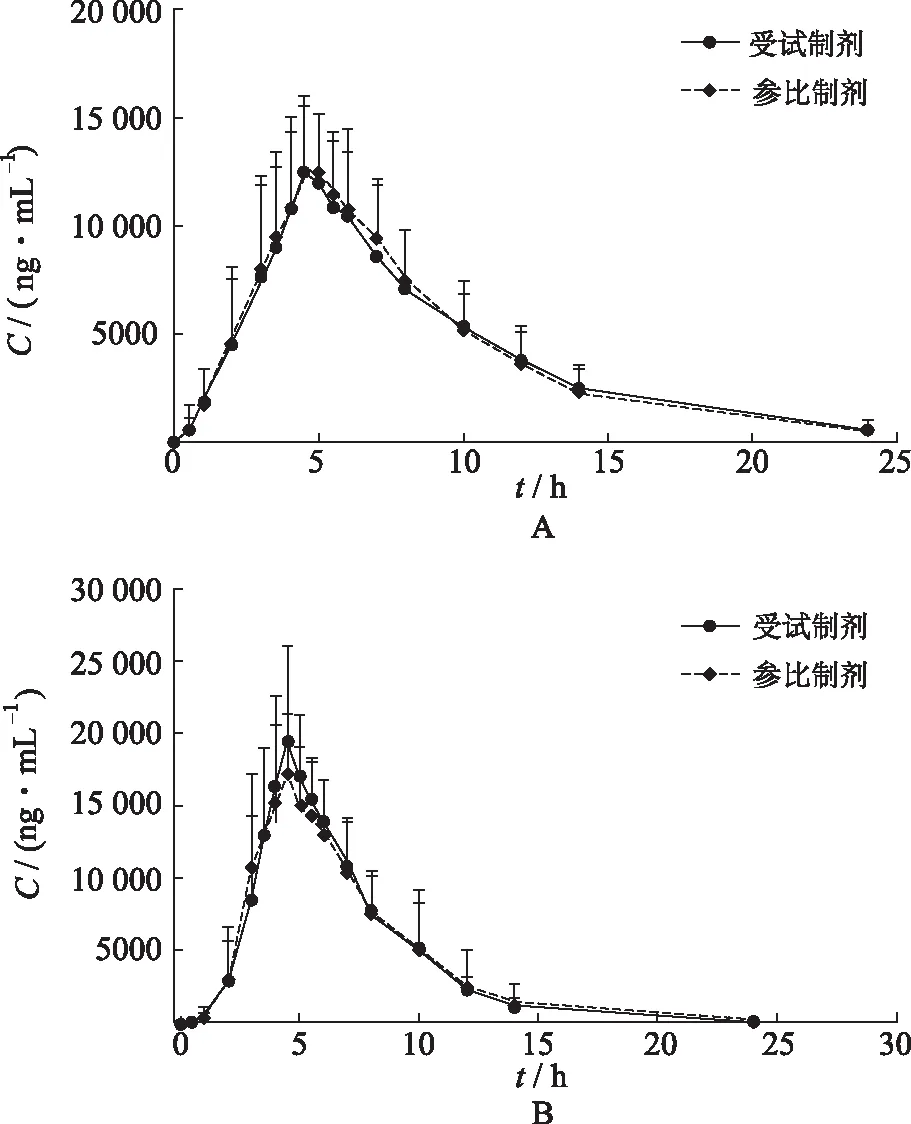
图3 空腹(A)和餐后(B)口服受试制剂和参比制剂平均血药浓度-时间曲线
2.6药动学参数 健康受试者空腹或餐后口服受试制剂和参比制剂后的药动学参数,见表2。

表2 健康受试者空腹和餐后条件下单次口服布洛芬缓释胶囊后PK参数
2.7生物等效性评价 空腹状态下,受试制剂和参比制剂给药后平均血药浓度-时间曲线高度相似,布洛芬tmax中位数均为4.5 h;在达到血药浓度峰值后,药物浓度下降,两种制剂中布洛芬的t1/2基本一致,受试制剂中布洛芬t1/2为(4.32±1.92) h,参比制剂中布洛芬t1/2为(4.10±1.23) h;将对数转换后的受试制剂和参比制剂的药动学参数Cmax、AUC0-t、AUC0-∞进行方差分析,采用双向单侧t检验及(1-2α)置信区间法进行生物等效性评价。结果表明,空腹试验中受试制剂与参比制剂主要药动学参数Cmax、AUC0-t、AUC0-∞几何均值比值的90%置信区间(CI)为(89.86%,102.08%),(94.05%,103.40%),(94.35%,104.72%)。
餐后状态下,受试制剂和参比制剂给药后平均血药浓度-时间曲线高度相似,布洛芬tmax中位数均为4.5 h;在达到血药浓度峰值之后,药物浓度下降,两种制剂中布洛芬的t1/2基本一致,受试制剂中布洛芬的t1/2为(2.97±0.34) h,参比制剂t1/2为(2.92±0.36) h;餐后试验中受试制剂与参比制剂主要药动学参数Cmax、AUC0-t、AUC0-∞几何均值比值的90%CI为(97.43%,112.29%),(95.44%,103.59%),(95.32%,103.62%)。
在空腹与餐后条件下,受试制剂与参比制剂主要药动学参数Cmax、AUC0-t、AUC0-∞几何均数比值的90%CI均在(80.00%,125.00%),满足生物等效性判定标准,表明健康成年受试者在空腹和餐后条件下单次口服受试制剂与参比制剂生物等效。
2.8安全性评价 空腹试验中服用受试制剂阶段发生不良事件6例次,受试者5例,发生率为20.8%(5/24),均为治疗期间的不良事件;与研究药物相关的不良事件6例次,受试者5例,发生率为20.8%(5/24);无导致退出试验的不良事件发生;无严重不良事件发生。服用参比制剂阶段不良事件发生11例次,受试者5例,发生率为20.8%(5/24),均为治疗期间的不良事件;与研究药物相关的不良事件6例次,受试者3例,发生率为12.5%(3/24);无导致退出试验的不良事件发生;无严重不良事件发生。所有不良事件严重程度均为轻度,1例不良事件结局为缓解,其余均为消失。
餐后试验中服用受试制剂阶段不良事件4例次,受试者4例,发生率为17.4%(4/23),均为治疗期间的不良事件;与研究药物相关的不良事件2例次,受试者2例,发生率为8.7%(2/23);无导致退出试验的不良事件发生;无严重不良事件发生。服用参比制剂阶段不良事件3例次,受试者2例,发生率为8.3%(2/24),均为治疗期间不良事件;与研究药物相关的不良事件2例次,受试者2例,发生率为8.3%(2/24);无导致退出试验的不良事件发生;无严重不良事件发生。所有不良事件严重程度均为轻度,所有不良事件的结局为消失。
3 讨论
国内报道布洛芬的检测方法多为HPLC法[3-4],服药剂量多为每次600 mg,加大给药剂量可能会增加胃肠道及其他不良反应的风险;本试验受试者服药剂量为300 mg,为临床用药提供可行的参考。本研究采用HPLC-MS/MS法测定人体血浆中布洛芬的浓度,该方法灵敏、准确可靠、快速简便,适用于布洛芬的血药浓度测定及其药动学和生物利用度研究。布洛芬缓释胶囊(芬必得®)药动学参数已有较多报道,梁俊等[5]在健康男性受试者中开展生物等效性研究,给予芬必得®300 mg后药动学参数分别为tmax:(4.9±1.1) h;Cmax:(13.6±5.8) μg·mL-1;AUC0-24:(95.9±45.2) μg·h·mL-1;AUC0-∞:(101.6±46.2) μg·h·mL-1。薛洪源等[6]开展单剂量及多剂量口服布洛芬缓释胶囊的药动学研究,给予芬必得®300 mg后药动学参数分别为tmax:(4.85±0.67) h;Cmax:(16.86±4.30) μg·mL-1;AUC0-24:(109.11±21.79) μg·h·mL-1;AUC0-∞:(109.11±22.92) μg·h·mL-1。DAVIES等[7]在布洛芬的临床药动学中提出,受试者分别于空腹或餐后条件下服用布洛芬800 mg可溶性骨架缓释片后,该制剂在药时曲线中呈现多个峰浓度,第一个峰浓度从空腹时14 mg·L-1升高至餐后时20 mg·L-1,差异有统计学意义,但空腹及餐后AUC差异无统计学意义。考虑到布洛芬属于BCSⅡ类药物,水中溶解度较低,高脂食物可以促进药物在胃肠道的溶解,因此药物的吸收速度会加快。此外由于布洛芬的口服生物利用度较高,因此食物对于吸收程度(AUC)无明显影响,试验结果与文献[5-7]报道较为一致。
安全性评价方面,口服受试制剂后不良事件为尿蛋白质定性弱阳性、总胆红素偏高、大便潜血弱阳性等。口服参比制剂后不良事件为凝血酶原时间延长、总胆红素偏高、大便潜血弱阳性、头晕等。受试制剂与参比制剂均无严重不良事件发生,未发生因不良事件而中止试验的情况。相较于既往文献报道,无明显差别[8]。本试验安全性评价结果表明,给予布洛芬缓释胶囊受试制剂与参比制剂300 mg后人体耐受性均良好,有较好的安全性,两制剂的不良事件发生情况也类似。
综上所述,布洛芬缓释胶囊受试制剂与参比制剂具有生物等效性,本试验结果可为我国布洛芬缓释胶囊生物等效性和药动学研究提供参考依据。
猜你喜欢
新传奇(2022年43期)2022-11-24
中国药学药品知识仓库(2022年5期)2022-04-11
口腔护理用品工业(2021年4期)2021-11-02
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
自我保健(2021年12期)2021-02-21
家庭医药(2019年7期)2019-07-25
中国科技纵横(2019年23期)2019-02-14
人生与伴侣·共同关注(2018年11期)2018-01-17
中国中药杂志(2016年23期)2017-04-07
海峡科技与产业(2017年1期)2017-03-04
