
ICP-MS法测定薄荷中13种重金属及有害元素含量的不确定度评定
2021-10-08 08:19朱雪妍林燕翔
中国民族民间医药 2021年15期
朱雪妍 罗 轶 林燕翔
广西壮族自治区食品药品检验所,广西 南宁 530021
薄荷为唇形科植物薄荷MenthahaplocalyxBriq.的干燥地上部分,其主要成分为挥发油等。目前《中国药典》2015年版一部仅对薄荷进行性状、鉴别、检查及挥发油的含量测定的检测,又中药材中的重金属已成为国际上对中国中药出口特别关注的问题之一,其安全性评价已经成为研究热点[1-2],故对于薄荷中重金属含量检测评价。本实验用电感耦合等离子体质谱法(ICP-MS)对薄荷的Cr、Mn、Co、Ni、Cu、As、Se、Mo、Cd、Ba、Dy、Tl、Pb等13种元素测定并对测定结果进行不确定度的评定。通过测量不确定度的评定,可定量评价测量结果的准确性,表示了测量的可信程度,指定了中药薄荷中的重金属及有害元素测量的相关影响因素,为正确评价和使用检验数据提供更科学更合理的依据,同时也对进一步的实验和研究引导了方向。
1 仪器与试剂
1.1 仪器 Agilent 8800 ICP―MS(美国Agilent公司); 雾化器为 Agilent 100μL min-1PFA Micro Flow Nebulizer,雾化室为石英双通道,Pilitier半导体控温于(2+1) ℃, 炬管为石英一体化,2.5 mm中心通道,采样锥材料为镍; Millipore Mill-Q 超纯水机, 美国Millipore Mill-Q 超纯水机(美国Millipore公司);CEM MARS6微波消解仪。
1.2 试剂 Ag、Al、As、Ba、Be、Ca、Cd、Co、Cr、Cs、Cu、Fe、Ga、K、Li、Mg、Mn、Na、Ni、Pb、Rb、Se、Sr、Tl、U、V、Zn混合标准液购于上海安普有限公司,浓度10 μg/mL,批号12-55YPY2;Mo单元素标准液购于中国计量科学研究院,规格 100μg/mL,批号14061;Dy单元素标准液购于上海安普有限公司,规格1000 μg/mL,批号1066825。
内标溶液为6Li、Sc、Ge、Y、In、Tb、Bi混合内标购于上海安普公司,规格10 μg/mL,12-16YPY2。调谐溶液为1μg/mL Ce、Co、Li、Mg、Tl、Y的混合溶液。
硝酸为微电子级(赛默飞世尔有限公司,批号1114100),水为经Millipore Milli-Q水处理系统处理后的去离子水。
1.3 样品 柑橘叶质控样购于中国地质科学院地球物理地球化学勘查研究所,批号GBW10020;薄荷叶实验用样品均购自于广西玉林药市。
1.4 仪器条件 经优化的ICP-MS参数:射频功率:1550 W;采样深度:10 mm;载气流速:1.07 L/min;He碰撞模式:He气流量 4.0 mL/min;积分时间:2S;测定时选取的同位素为52Cr、55Mn、59Co、60Ni、63Cu、75As、78Se、95Mo、114Cd、137Ba、163Dy、205Tl、208Pb,其中52Cr、55Mn以45Sc为内标,59Co、60Ni、63Cu使用仪器计算的虚拟内标,75As、78Se、95Mo以72Ge为内标,114Cd以115In为内标,137Ba、163Dy以159Tb为内标,205Tl、208Pb以209Bi为内标。
2 样品制备
2.1 供试品溶液、空白溶液、质控样品溶液制备 取薄荷样品粉碎成粗粉,取约0.2 g,精密称定,置消解罐中,加硝酸溶液8 mL、氢氟酸0.5 mL,混匀,置微波消解仪中消解。采用程序升温,功率1030~1800 W,爬升时间20~25 min,190 ℃保持20 min。消解完毕后放冷,置赶酸架上130 ℃赶酸2.5 h,放冷至室温,转入25 mL量瓶中,用2%硝酸溶液洗涤消解罐3~4次,并稀释至刻度,摇匀,即得。同法同时制备样品空白溶液及质控样溶液。
2.2 内标溶液制备 取混合内标溶液用2%硝酸溶液稀释50倍,即得。
2.3 标准溶液制备
2.3.1 混合标准储备液 分别精密吸取混合标准溶液(10 μg/mL)、Mo(100 μg/mL)及Dy(1000 μg/mL)适量,用2%硝酸溶液配制成含元素均为Cr、Mn、Co、Ni、Cu、Se、Mo、Cd、Ba、Dy、Tl、Pb1000 ng/mL及200 ng/mL的标准储备溶液。
再精密吸取混合标准溶液(10 μg/mL)适量,用2%硝酸溶液配制成500 ng/mL溶液作为As测定的标准储备溶液。
2.3.2 绘制标准曲线 分别精密量取混合标准储备液适量,分置25 mL、50 mL、100 mL的容量瓶中,配制成Cr、Mn、Co、Ni、Cu、Se、Mo、Cd、Ba、Dy、Tl、Pb浓度分别为0.1 ng/mL、0.2 ng/mL、1 ng/mL、2 ng/mL、10 ng/mL、50 ng/mL、100 ng/mL的标准溶液与混合储备溶液200 ng/mL、1000 ng/mL共同作为系列标准溶液及As浓度为0.5 ng/mL、2.5 ng/mL、12.5 ng/mL、50 ng/mL、100 ng/mL的系列标准溶液。
选取各元素与样品浓度相适应的点(详见5.2),用比率与浓度进行线性回归。
结果十三种元素的线性关系良好,相关系数均在0.9998以上。
2.4 质控物质对照 采用国家标准物质柑橘叶作为随行工作对照物质,用以检测方法的可行性,13种元素的测定值均符合要求。

表1 标准曲线方程

续表1
2.5 重复性试验 取薄荷样品0.2g,6份,按“3.1”项方法制备,按“2”项条件测定,所得各元素的含量为52Cr 5.88 mg/kg、55Mn 47.2 mg/kg、59Co 0.416 mg/kg、60Ni 1.17 mg/kg、63Cu 12.4 mg/kg、75As 2.01 mg/kg、78Se 0.110 mg/kg、95Mo 0.345 mg/kg、114Cd 0.1252 mg/kg、137Ba 20.61 mg/kg、163Dy 0.146 mg/kg、205Tl 0.0353 mg/kg、208Pb 3.43 mg/kg。
2.6 加样回收试验 取上述重复性试验所用的薄荷样品0.2g,6份,加入适当量的标准溶液,按“3.1”项方法制备,按“2”项条件测定,计算回收率,各元素的加样回收均在80%~125%之间,符合痕量分析的要求。

表2 加样回收率
3 数学模型及不确定度来源分析[3]
P:单位质量样品被测元素含量(mg/kg);C0:仪器测试用的样液浓度(mg/kg);V:样品定容体积(mL);f:稀释因子(本实验中为1);m:样品重量(g)
由数学模型及实验过程可知引入不确定度的因素由图1[3]可见:引入不确定度的主要有样品称重、样品的前处理、定容体积等原因。

图1 引入不确定度的因素图
4 实验分析及数据处理
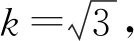
4.2 多份样品的重复性引入的不确定度up,rel对薄荷样品取6份,同时实验测定。按A类不确定度评定[3],
c0:样品的含量的平均值;S:样品的标准偏差;n:样品重复性测定的次数;

表3 重复性引入的不确定度
4.3 供试品溶液制备过程中引入的相对标准不确定度uR,rel由于样品处理消解过程中受基体效应及消解方法影响,供试品中加入的元素不能100%进入溶液中进行测定。按照显著性检验,若平均回收率与1.0有显著性差异,引入的不确定度即用平均回收率的标准偏差表示[5]。


薄荷测定的各元素的回收率的不确定度为(本实验中,查表得tcrit=2.57)
因元素Cr、 Mn 、As 、Pb的回收率的不确定度与100%无显著性差异,即可认为消解制备过程较为完全,不引入不确定度的变化。
4.4 由定容引入的不确定度u(v)样品消解液定容至25 mL 容量瓶中,其引入的不确定度u(25mL)由容量瓶的校准引入的u1(25mL)、温度效应引入的u2(T,25mL)及体积重复性引入的u3(25mL)合成[6]:

续表4

表4 回收率引入的不确定度


其相对标准不确定度u1(25mL)为0.0173/25 mL=0.000693。
4.4.2 温度效应引入的相对标准不确定度
V:容量瓶或移液器体积(mL);ΔT:实验室实际温度与实验室标准温度20℃的偏差(本实验室实际温度为25℃);2.1×10-4:水的膨胀系数,单位:℃-1。

4.4.3 体积重复性产生的相对标准不确定度u3(25mL)对25mL容量瓶用水定容至刻度,称重,重复10次,计算其标准偏差S=0.010,即
u3(25mL)=S/25=0.000401。
5 样品溶液中元素浓度引起的不确定度
uxo,rel:样品浓度的合成相对不确定;uB,rel:标准溶液配制引入的相对不确定度;uc,rel:标准曲线拟合引入的相对不确定度。
5.1.1 标准物质引入的不确定度ub,rel=U/K 由标准物质证书中查得标准液扩展不确定度U(%)及扩展因子(k=2)得,所引入的不确定度ub,rel为Cr、Mn、Co、Ni、Cu、As、Se、Cd、Ba、Tl、Pb的ub,rel=0.5%/2=0.0025;Mo的ub,rel=1%/2=0.005;、Dy的ub,rel=0.3%/2=0.0015。
u(v1)、u(v2)…u(vn):体积分别为v1、v2、…vn的容量瓶或移液器引入的相对不确定度,n1、n2、…nn:体积分别为v1、v2、…vn容量瓶或移液器在标准溶液的稀释过程中使用的次数,由容量瓶及移液器引入的不确定度u(v)包括校准引入的不确定度u1(v)、温度效应引入的不确定度u2(T,v)、定容溶液体积(移液器吸取体积)引入的不确定度u3(v).
校准的不确定度:实验中使用的100 mL、50 mL及25 mL的容量瓶,0.05 mL、0.1 mL、0.5 mL、1.00 mL、2.5 mL、5.00 mL的移液器,A级的最大允差分别为±0.1 mL、±0.05 mL、±0.03 mL及3%、2%、1%、1%、0.5%、0.6%。按矩形分布,即各容量瓶及移液器的相对标准不确定度u1(v)为
温度效应引起的不确定度见4.4.2;各容量瓶及移液器引入的不确定度均为0.000606。
体积重复性引入的不确定度:对各容量瓶及移液器用水定容或吸取至刻度,称重,重复10次,计算其标准偏差S(V)。其相对标准不确定度u3(v)=S(V)/V
各容量瓶及移液器引入的u(v)见表5~6。

表5 容量瓶引入的不确定度

表6 移液器引入的不确定度
由稀释因子n引入的不确定度:根据标准曲线配制过程中各元素所需要的容量瓶及移液器的次数,计算各元素的uD,rel
5.1.3 各元素在标准溶液配制过程中引入的合成相对标准不确定度uB,rel计算结果见表7。

表7 标准曲线配制过程引入的不确定度

续表7
5.2 标准曲线拟合的不确定度urel,C校正曲线法采用最小二乘法拟合成标准曲线,拟合曲线有关的不确定度由下式表示[8]:

SR:标准曲线的剩余标准差;B:斜率;n:标准曲线点数;p:待测样品测定次数;C0:回归曲线各点浓度的平均值;C:待测样品的平均值;C0j:各标准曲线的浓度;Aaj:各标准曲线的实际响应值;Aj:根据回归曲线计算的理论值uc:标准曲线拟合的标准不确定度。
5.3 样品溶液引入的合成相对标准不确定度uxo,rel
各元素相对标准不确定度的计算[9]结果如表9。

表9 供试品溶液中的元素浓度合成不确定度计算

续表8

表8 各元素标准曲线拟合引入的不确定度
5.4 各元素扩展不确定度和结果报告
各元素总的合成相对不确定度为:
u(c)=uc,rel×c;U=k×u(c)
取包含因子k=2,对应的置信概率为95%,计算扩展不确定度U[10],同时报告结果,见表10。

表10 各元素的扩展不确定度及报告结果
6 讨论
由测定结果和计算可知,薄荷测量不确定度的主要来源为加样回收率、多份样品的重复性及样品溶液浓度引入的不确定。取样称重,供试品定容等引入的不确定度与上述几项相比,均在最大不确定度分量十分之一以下,在最后的合成计算中可以忽略不计。在以后的相关方法的不确定度的讨论中,可以不考虑此两项因素的影响。实验前处理消解更完全,实验测定就更精准。分析实验数据得,标准曲线配制所得的浓度越接近理想值,标准曲线拟合产生的不确定度也会更小,溶液浓度的测定也更精准,提高标准曲线的配置水平极其重要。同时持续受控测量过程,保持测量结果的稳定一致,特别关注加样回收率、重复性、样品溶液浓度和标准曲线因素,及时对测量的影响做修正。
7 结论
重金属及有害元素测定属于痕量分析,其不确定度的研究是分析出影响测量值和真实值偏差的因素,从而降低方法中的不确定度,让实验数据更准确可靠的必不可少途径。经评定探讨,实验室减少不确定度引入方案如下:调整前处理方法,应对不同类的样品使用与其相适宜的消解溶剂及程序方案;维护仪器良好的工作状态,提高标准曲线配制方法和水平,保证样品溶液于对照品溶液的基体、浓度的匹配;提高检验人员在整个实验过程中操作水平,及细致敏感度。
猜你喜欢
中国动物保健(2022年2期)2022-05-05
商品与质量(2021年28期)2021-07-22
中国应急管理科学(2021年4期)2021-04-13
读友·少年文学(清雅版)(2020年6期)2020-10-20
读友·少年文学(清雅版)(2020年4期)2020-08-24
读友·少年文学(清雅版)(2020年2期)2020-06-15
读友·少年文学(清雅版)(2020年1期)2020-05-20
技术与市场(2020年5期)2020-03-02
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
