
阿嗪米特中潜在基因毒性杂质马来酰肼的含量测定
2021-09-28 01:09赵钰馨孙秉喆倪卫星梁然植狄斌苏梦翔
中国药房 2021年18期
关键词:高效液相色谱
赵钰馨 孙秉喆 倪卫星 梁然植 狄斌 苏梦翔

中圖分类号 R917 文献标志码 A 文章编号 1001-0408(2021)18-2189-05
DOI 10.6039/j.issn.1001-0408.2021.18.03
摘 要 目的:建立阿嗪米特原料药中潜在基因毒性杂质马来酰肼含量测定的方法。方法:采用高效液相色谱-荧光检测器法。以Thermo Syncronis C18为色谱柱,以0.2 mol/L乙酸溶液-甲醇为流动相进行梯度洗脱;柱温为30 ℃;激发波长为315 nm,发射波长为389 nm;流速为1 mL/min;进样量为20 μL。结果:空白溶剂和阿嗪米特均不干扰马来酰肼的测定;马来酰肼检测质量浓度的线性范围为19.5~300 ng/mL(r=0.999 9);检测限为4.5 ng/mL,定量限为19.5 ng/mL;加样回收率为98.79%~103.76%(RSD均小于3.00%,n=9);精密度、稳定性(24 h)试验的RSD均不大于5.63%,且耐用性试验的RSD均小于2.00%(n=6)。3批阿嗪米特原料药中均未检出马来酰肼。结论:该方法专属性好、灵敏度高、定量准确,可用于阿嗪米特或其他基质中马来酰肼的痕量测定。
关键词 马来酰肼;阿嗪米特原料药;高效液相色谱-荧光检测器法;基因毒性杂质
Content Determination of Potential Genotoxic Impurity Maleic Hydrazide in Azintamide
ZHAO Yuxin1,SUN Bingzhe1,2,NI Weixing2,LIANG Ranzhi2,DI Bin1,3,4,SU Mengxiang1,3,4[1. School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China; 2. Yangzhou Yiyang Pharmaceutical Co., Ltd., Jiangsu Yangzhou 225600, China; 3. Key Laboratory of Drug Quality Control and Pharmacovigilance (China Pharmaceutical University), Ministry of Education, Nanjing 210009, China; 4. Key Laboratory for Research and Evaluation of Pharmaceutical Preparations and Excipients, National Medical Products Administration, Nanjing 210009, China]
ABSTRACT OBJECTIVE: To establish a method for the content determination of potential genotoxic impurity maleic hydrazide in azintamide raw material. METHODS: HPLC-FLD method was adopted. The determination was performed on Thermo Syncronis C18 column with mobile phase consisted of 0.2 mol/L acetic acid-methanol (gradient elution). The column temperature was set at 30 ℃, the excitation wavelength was 315 nm and emission wavelength was 389 nm. The flow rate was 1 mL/min, and the sample size was 20 μL. RESULTS: The blank solvent and azintamide did not interfere with the determination of maleic hydrazide. The linear range of maleic hydrazide was 19.5-300 ng/mL (r=0.999 9). The limit of detection was 4.5 ng/mL and the limit of quantification was 19.5 ng/mL. The recovery ranged from 98.79% to 103.76% (RSDs were lower than 3.00%,n=9). RSDs of precision and stability(24 h)tests were no more than 5.63%,and those of durability tests were less than 2.00% (n=6). Maleic hydrazide was not detected in 3 batches of azinamide raw material. CONCLUSIONS: The method is specific, sensitive and accurate. It can be used for the trace determination of maleic hydrazide in azintamide or other matrix.
KEYWORDS Maleic hydrazide; Azintamide; HPLC-FLD; Genotoxic impurity
马来酰肼(maleic hydrazide)的化学名为顺丁烯二酰肼,是一种合成哒嗪类化合物的常用原料。哒嗪类化合物作为重要的中间体,被广泛应用于农药及头孢唑兰、普纳替尼等药品的合成中[1-4]。尽管目前尚无证据证明马来酰肼具有基因毒性和致癌性,但其结构中存在警示结构——肼,故该成分为潜在的基因毒性杂质[5]。潜在基因毒性杂质的毒性虽然未经实验证实或模型验证,但人用药品注册技术要求国际协调会、美国FDA及欧洲药品管理局等均对其在药品中的限度提出了具体要求[6-8]。
阿嗪米特(azintamide)的化学名为2-(6-氯哒嗪-3-基)硫烷基-N,N-二乙基乙酰胺,于1959年首次合成[9],是一种促进胆汁分泌的药物,可用于治疗因胆汁分泌不足而引起的消化系统方面的疾病[10-11]。目前,阿嗪米特已成为临床上主要使用的消化酶类药物之一[11],而马来酰肼为其合成的起始物料[12]。因此有必要建立灵敏度高、专属性强、基质干扰小、重复性好的痕量定量分析方法来对马来酰肼在阿嗪米特原料药中的残留量进行考察。由于马来酰肼沸点高、不易气化,常用的检测方法有高效液相色谱(HPLC)-紫外法(检测限135 ng/mL)、液质联用法(检测限0.5 ng/mL)等[13-15]。其中,液质联用技术的检测限虽然可达0.5 ng/mL[15],但易受大型仪器设备成本高和普及性低的限制。鉴于此,本研究利用马来酰肼的荧光特性,采用选择性好、灵敏度高的高效液相色谱-荧光检测器法(HPLC-FLD)对阿嗪米特原料药中潜在基因毒性杂质马来酰肼进行定量分析,以期为其质量控制提供参考。
1 材料
1.1 主要仪器
RF-6000型荧光分光光度计、LC-20AD型HPLC仪(包括RF-20A型FLD、LabSolutions 6.81网络版工作站)均购自日本Shimadzu公司;BT25S型电子天平购自德国Sartorius公司;Synergy UV型超纯水系统购自德国Merck公司;KH-250DB型数控超声波清洗器购自昆山禾创超声仪器有限公司。
1.2 主要药品与试剂
马来酰肼和阿嗪米特的结构式见图1。
马来酰肼对照品(美国AccuStandard公司,批号02317JT,纯度100%);阿嗪米特原料药(由扬州一洋制药有限公司提供,由韩国东邦化学株式会社生产,批号分别为190011、190012、190013,纯度均不低于99%);乙酸(南京化学试剂股份有限公司)为分析纯,甲醇(美国Tedia公司)为色谱纯,水为超纯水。
2 方法与结果
2.1 色谱条件
以Thermo Syncronis C18(250 mm×4.6 mm,5 μm)为色谱柱,以0.2 mol/L乙酸溶液(A)-甲醇(B)为流动相进行梯度洗脱(0~3 min,90%A;3~4 min,90%A→10%A;4~12 min,10%A;12~13 min,10%A→90%A;13~18 min,90%A);柱温为30 ℃;激发波长为315 nm,发射波长为389 nm;流速为1 mL/min;进样量为20 μL。
2.2 溶液的制备
2.2.1 对照品溶液 取马来酰肼对照品约20 mg,精密称定,置于20 mL量瓶中,加溶剂[0.2 mol/L乙酸溶液-甲醇(1 ∶ 1,V/V),下同]适量,超声(频率40 kHz,功率250 W,下同)处理30 min使溶解,然后加溶剂稀释至刻度,摇匀,制成质量浓度为1 mg/mL的对照品贮备液。取上述对照品贮备液30 μL,置于10 mL量瓶中,加溶剂稀释至刻度,摇匀,制成每1 mL含马来酰肼3 μg的对照品溶液。
2.2.2 限度对照品溶液 取“2.2.1”项下对照品溶液500 μL,置于10 mL量瓶中,加溶剂稀释至刻度,摇匀,制成每1 mL含马来酰肼150 ng的限度对照品溶液。
2.2.3 含限度浓度杂质的供试品溶液 取阿嗪米特原料药约500 mg,精密称定,置于10 mL量瓶中,加溶剂适量,然后加入“2.2.1”项下对照品溶液500 μL,超声处理30 min使溶解,然后加溶剂稀释至刻度,摇匀,制成每1 mL含马来酰肼150 ng的含限度浓度杂质的供试品溶液。
2.2.4 供试品溶液 取阿嗪米特原料药约500 mg,精密称定,置于10 mL量瓶中,加溶剂适量,超声处理30 min使溶解,然后加溶剂稀释至刻度,摇匀,即得供试品溶液。
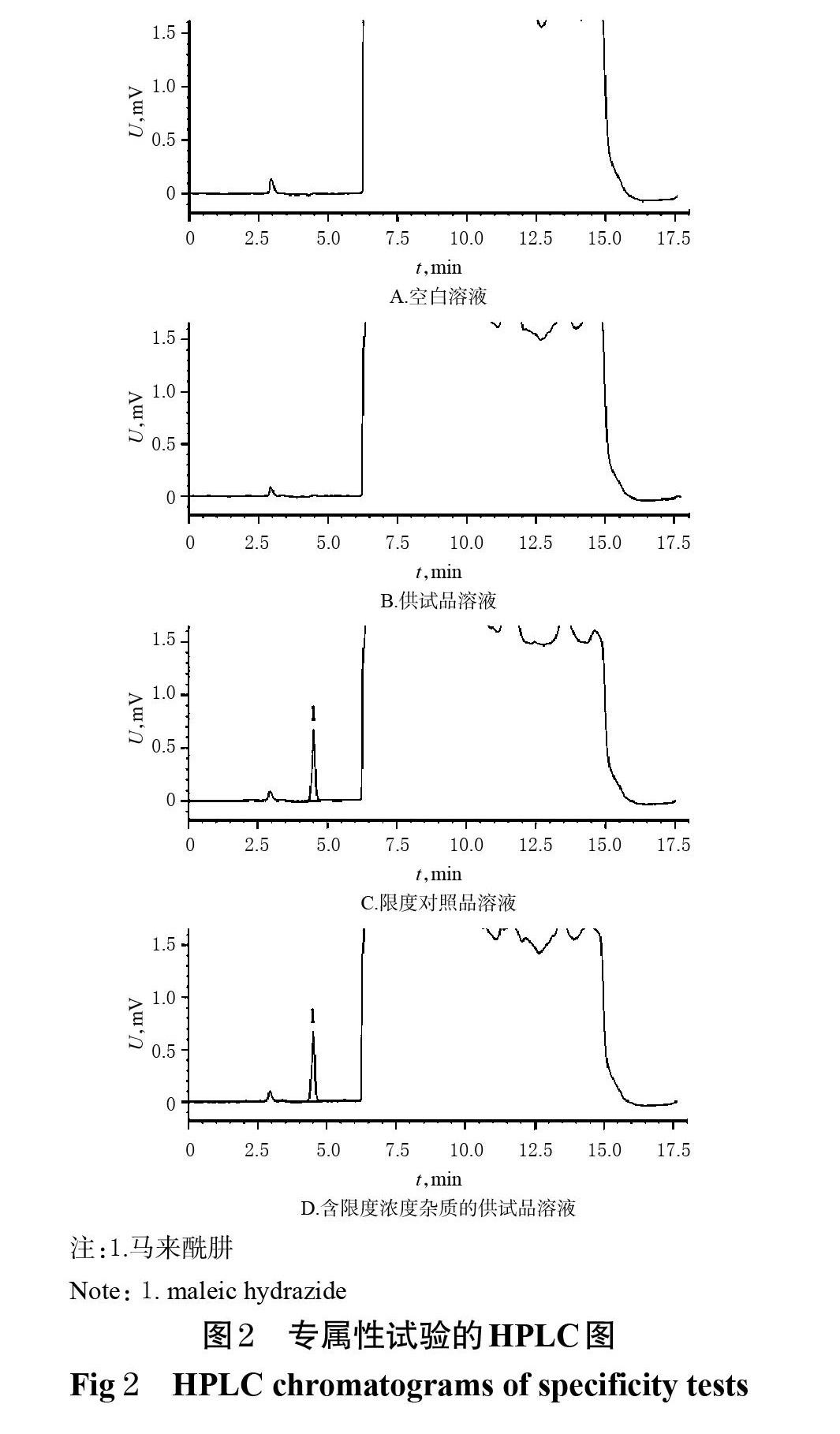
2.3 专属性试验
取空白溶液(溶剂)以及“2.2”项下限度对照品溶液、供试品溶液和含限度浓度杂质的供试品溶液各20 μL,分别按“2.1”项下色谱条件进样测定,记录色谱图。结果显示,马来酰肼的保留时间在4.5 min左右,其色谱峰峰形良好,基线平稳;理论板数以马来酰肼计大于6 000,马来酰肼色谱峰与相邻色谱峰间的分离度大于1.5,且空白溶液不干扰测定。专属性试验的HPLC图见图2。
2.4 线性关系考察
分別取“2.2.1”项下对照品溶液65、150、250、500、750、1 000 μL,置于不同10 mL量瓶中,加溶剂稀释至刻度,制成马来酰肼质量浓度分别为19.5、45、75、150、225、300 ng/mL的系列标准溶液。取上述系列标准溶液,分别按“2.1”项下色谱条件进样测定,记录峰面积。以峰面积(y)为纵坐标、待测物质量浓度(x,ng/mL)为横坐标,采用最小二乘法进行线性回归。结果显示,马来酰肼的线性方程为y=34.709x+10.472(r=0.999 9),该成分检测质量浓度的线性范围为19.5~300 ng/mL(相当于限度的13%~200%)。
2.5 检测限和定量限考察
取“2.2.2”项下限度对照品溶液适量,用溶剂逐级稀释,然后分别按“2.1”项下色谱条件平行测定6次。将测得的信号与空白处的信号(基线噪音)进行比较,分别以信噪比3 ∶ 1、10 ∶ 1计算检测限和定量限。结果,马来酰肼的检测限为4.5 ng/mL、定量限为19.5 ng/mL,6次平行测定的定量限的RSD为4.03%(n=6),表明該方法灵敏、稳定。
2.6 精密度试验
2.6.1 仪器精密度 取“2.2.3”项下含限度浓度杂质的供试品溶液,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果显示,杂质保留时间和峰面积的RSD分别为0.05%、0.74%(n=6),表明仪器精密度良好。
2.6.2 重复性 取阿嗪米特原料药(批号190011)适量,按“2.2.3”项下方法制备含限度浓度杂质的供试品溶液,平行制备6份。分别按“2.1”项下色谱条件进样测定,记录峰面积并按外标法计算杂质的含量。结果显示,杂质含量的RSD为2.69%(n=6),表明方法的重复性良好。
2.6.3 中间精密度 分别由2名实验人员于不同时间点按“2.2.3”项下方法制备含限度浓度杂质的供试品溶液(批号190011),各平行制备6份。分别按“2.1”项下色谱条件进样测定,记录峰面积并按外标法计算杂质含量。结果显示,2名实验人员测得杂质含量的RSD为5.63%(n=12),表明方法的中间精密度良好。
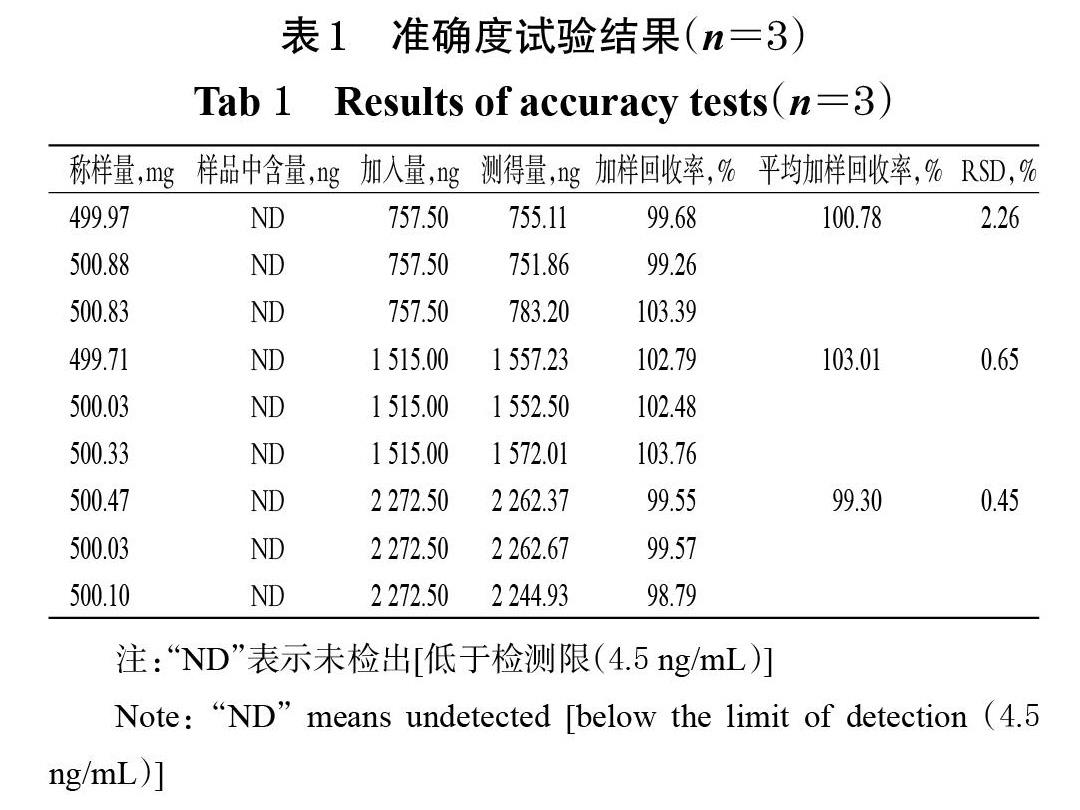
2.7 准确度试验
取阿嗪米特原料药(批号190011)约500 mg,共9份,精密称定,分别置于10 mL量瓶中,加溶剂适量,然后分别加入“2.2.1”项下对照品溶液250、500、750 μL,各3份,超声处理30 min使溶解,加溶剂稀释至刻度,摇匀,分别作为质量浓度相当于杂质限度50%、100%和150%的供试品溶液。取上述溶液,分别按“2.1”项下色谱条件进样测定,记录峰面积,按外标法计算马来酰肼的含量并计算其加样回收率。结果显示,各质量浓度供试品溶液中马来酰肼的加样回收率在98.79%~103.76%范围内,RSD均小于3.00%(n=3),表明方法的准确度良好。准确度试验结果见表1。
2.8 稳定性试验
取“2.2.2”项下限度对照品溶液和“2.2.3”项下含限度浓度杂质的供试品溶液,分别于室温下放置0、1、2、3、4、5、6、7、8、9、10、11、12、24 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果显示,限度对照品溶液和含限度浓度杂质的供试品溶液中马来酰肼峰面积的RSD分别为1.55%和1.62%(n=14),表明上述溶液在室温下放置24 h稳定。
2.9 耐用性试验
按“2.2.3”项下方法制备含限度浓度杂质的供试品溶液(批号190011),在其他色谱条件不变的情况下,采用外标法分别考察流动相A不同浓度(0.196 7、0.200 0、0.203 3 mol/L)、不同流速(0.9、1.0、1.1 mL/min)、不同柱温(25、30、35 ℃)、不同检测波长[激发波长、发射波长(313 nm、387 nm;315 nm、389 nm;317 nm、391 nm)]对含量测定结果的影响,并计算其回收率,每个条件平行测定2次,取平均值。结果显示,各种条件下马来酰肼回收率的RSD均小于2.00%(n=6),表明方法的耐用性良好。耐用性试验结果见表2。
2.10 样品中杂质含量测定
取3批阿嗪米特原料药,分别按“2.2.4”项下方法制备供试品溶液,然后按“2.1”项下色谱条件进样测定,记录峰面积并按外标法计算马来酰肼含量。结果显示,3批阿嗪米特原料药中均未检出马来酰肼。阿嗪米特中马来酰肼含量测定结果见表3。
3 讨论
3.1 限度的确定
马来酰肼作为3类致癌物质,尚无充足证据证实其致癌性,但其对非目标生物的慢性毒性仍存在不确定性[15]。由于马来酰肼具有潜在的基因毒性,故本研究按基因毒性杂质分类中的2类物质使用毒理学关注阈值(TTC)对其进行限度控制[8],以确保药品的安全性[16-17]。根据TTC计算可接受摄入量发现,一个具有诱变性的杂质每天每人摄入1.5 μg时其风险被认为是可以忽略的(终生暴露情况下,理论的患癌风险小于十万分之一)[18]。基因毒性杂质可接受限度(ppm)=TTC限度(μg/d)/药物的最大日剂量(g/d)[6]。根据阿嗪米特说明书中每天最大剂量450 mg,可计算出马来酰肼的可接受限度(ppm)=1.5(μg/d)/0.45(g/d)=3.33 ppm。因此,笔者将马来酰肼的限度定为3 ppm。本研究所建立方法的检测限可达0.09 ppm(4.5 ng/mL),远低于上述限度。通过本研究建立的方法对3批阿嗪米特原料药进行测定,结果均未检出马来酰肼,表明3批原料中马来酰肼的含量均低于检测限,满足限度控制的要求。
3.2 检测方法的选择
笔者前期试验发现,采用HPLC-紫外法进行测定的灵敏度不能满足检测要求。因本品含有共轭结构,具有荧光响应,考虑FLD的专属性和灵敏度均优于紫外检测器,所以笔者最终采用了HPLC-FLD法进行检测。马来酰肼是弱酸,在不同pH条件下其电离平衡会发生改变,荧光强度也会因其离解状态不同而发生改变[19]。笔者在预试验中发现,当流动相为0.1 mol/L乙酸溶液和甲醇时,马来酰肼的响应信号较弱,无法满足测定需要;当降低流动相pH后,马来酰肼的响应信号明显增强,故本研究最终确定流动相为0.2 mol/L乙酸溶液和甲醇。此外,笔者还通过固定激发波长对马来酰肼对照品溶液进行发射波长扫描以及固定发射波长进行激发波长扫描,反复调整优化后确定激发波长为315 nm、发射波长为389 nm。本研究建立的HPLC-FLD法与Kubilius等[14]建立的方法(检测限25 ng/mL)相比,检测限更低(可达4.5 ng/mL)、峰形更理想。
综上所述,本研究以阿嗪米特原料药为检测样品,成功建立了测定马来酰肼含量的HPLC-FLD法。方法学考察结果表明,该方法专属性好、灵敏度高、定量准确,可为阿嗪米特或其他药品中马来酰肼的痕量测定提供参考。但此方法中流动相的水相为0.2 mol/L乙酸溶液,酸性较强,对色谱柱有一定损伤。此外,本研究仅检验了3批阿嗪米特原料药,样本量较少,有待进一步扩大样本量加以研究。
参考文献
[ 1 ] 罗维,苑丽红,李保庆,等.新型含哒嗪环的1,3,4-噻二唑类化合物的合成及杀菌活性[J].农药,2016,55(10):721-724.
[ 2 ] 唐武,王鹏旭,金波,等. 3-炔基咪唑并[1,2-b]哒嗪的简便合成[J].合成化学,2016,24(7):643-646.
[ 3 ] 陈春光,冯亚青,陈学玺.咪唑并[1,2-b]哒嗪的合成[J].精细化工,2012,29(8):783-786.
[ 4 ] 程伟,张向龙,陈启绪,等. 3-碘-6-氯咪唑并[1,2-b]哒嗪的合成研究[J].化学试剂,2020,42(12):1492-1496.
[ 5 ] MATVEEVA O A,KOVALEVA E L. Modern approaches to estimating the content of genotoxic impurities in drugs:a review[J]. Pharm Chem J,2016,49(11):765-770.
[ 6 ] 汪生,杭太俊.药物中基因毒性杂质检测策略的研究[J].中国新药杂志,2019,28(23):2840-2846
[ 7 ] 谢含仪,林云良,张瑞凌,等.基因毒性杂质分析方法和前处理技术的研究进展[J].药物分析杂志,2018,38(10):1668-1676.
[ 8 ] ICH. ICH M7(R2):assessment and control of DNA re- active(mutagenic)impurities in pharmaceuticals to limit potential carcinogenic risk[S]. 2018-09-19.
[ 9 ] ABDEL-MOETY E M,AL-KHAMEES H A. Analytical profiles of drug substances:volume 18[M]. California: Academic Press,1989:1-31.
[10] HEGAZY M A,HASSANAIN W A,ABDEL FATTAH L E,et al. Chromatographic study of azintamide in bulk powder and in pharmaceutical formulation in the presence of its degradation form[J]. J AOAC Int,2017,100(2):422-428.
[11] 苑藝,郭婉婕,韩晟.复方阿嗪米特肠溶片和复方消化酶胶囊治疗胆系相关消化不良的药物经济学评价[J].中国新药杂志,2020,29(10):1195-1200.
[12] 江西博雅欣和制药有限公司.一种制备阿嗪米特的方法:中国,201810328849.X[P].2018-06-29.
[13] 吕小刚,顾丽莉,孔光辉,等.超临界萃取-HPLC法检测烟叶中马来酰肼残留[J].精细化工,2016,33(12):1370- 1374.
[14] KUBILIUS D T,BUSHWAY R J. Determination of ma- leic hydrazide in potatoes and onions by fluorescence high performance liquid chromatography[J]. J Liq Chromatogr R T,1999,22(4):593-601.
[15] ABDELWAHED M H,KHORSHED M A,ELMARSAFY A M,et al. Polar reversed-phase liquid chromatography coupled with triple quadrupole mass spectrometer method for simple and rapid determination of maleic hydrazide residues in some fruits and vegetables[J]. Food Anal Method,2021,14(1):172-185.
[16] 杨莉,梅勇,龙涛,等. HPLC法测定对乙酰氨基酚片中有关物质的含量[J].中国药房,2020,31(10):1233-1238.
[17] 刘克锋,刘宇,王雪芹,等. LC-MS/MS法同时测定艾司奥美拉唑钠原料药及其制剂中3种杂质的含量[J].中国药房,2019,30(6):775-779.
[18] 毛颐晴,李纬,朱丽君. GC-MS法测定甲磺酸萘莫司他原料药中的遗传毒性杂质[J].中国药房,2016,27(18):2555-2557.
[19] 许金钩,王尊本.荧光分析法[M].北京:科学出版社,2006:53-54.
(收稿日期:2021-05-01 修回日期:2021-07-21)
(编辑:林 静)
猜你喜欢
分析化学(2017年1期)2017-02-06
中国医药导报(2016年30期)2016-12-28
热带农业科学(2016年10期)2016-12-12
分析化学(2016年7期)2016-12-08
上海医药(2016年21期)2016-11-21
科学与财富(2016年28期)2016-10-14
上海医药(2016年3期)2016-03-23
现代仪器与医疗(2015年4期)2015-07-15
