
基于FF和VIM的嗜热蛋白质预测*
2021-09-27 05:00王鲜芳刘依锋李启萌
河南工学院学报 2021年4期
王鲜芳,卢 凡,刘依锋,李启萌
(1.河南工学院 计算机科学与技术学院,河南 新乡 453003;2.河南师范大学 计算机与信息工程学院,河南 新乡 453007)
0 引言
嗜热蛋白质在开发应用方面有着非常广阔的前景。在发酵行业中,可利用其能耐受高温这一特性,提高反应温度和速度,减少中温型杂菌污染的机会[1],来生产多种催化剂。由嗜热蛋白质生产的这些酶制剂具有热稳定性好,催化反应速率高,易于在室温下保存等优点。嗜热蛋白质研究中最引人注目的成果之一是将水生栖热菌中耐热的TaqDNA聚合酶[2]用于基因研究和遗传工程以及基因技术之中。所以对于嗜热蛋白质的预测显得尤为重要。
目前嗜热蛋白质预测主要有两类方法:生物实验手工标注和计算方法预测[3]。在后基因组时代,随着DNA和蛋白质序列以及结构信息的大量积累,传统的手工实验费时费力,所以人们更倾向于利用数学、计算机科学知识分析、挖掘生物数据,以寻求蕴含在其中的生物规律[4]。近年来,机器学习算法被广泛应用到蛋白质预测中,目前常用的机器学习分类算法有线性判别分析、K近邻算法、决策树、朴素贝叶斯、支持向量机[5]和神经网络等。其中,Hua和Sun[6]等人采用氨基酸组成方法构造特征表达模型,在蛋白质数据集上取得了高达91.4%的预测准确率;Wu[7]等人运用决策树预测嗜热蛋白质的热稳定性,其精度达到80%以上;Dao[2]等人运用支持向量机对嗜热蛋白质进行预测,在五折叠交叉验证中精度达到了92.75%。以上实验都取得了较好的预测精度,但是预测性能还需要进一步提升,同时这些实验都为我们进一步提升[8]嗜热蛋白质的预测精度提供了很好的借鉴。
现有的预测研究方法大部分只是提取单一的蛋白质特征,并未全面地将嗜热蛋白质的内在信息表达出来,在一定程度上限制了模型的预测性能。为此,本文提出了基于特征融合(Feature Fusion,FF)的多层感知机(Multi-Layer Perception,MLP)预测模型。首先,分别使用多段氨基酸组成(Multi-stage Amino Acid Composition,MAAC)、G-gap二肽组成和组成转变分布组成(Composition Transition Distribution Composition,CTDC)来表征嗜热蛋白质序列,通过串联融合的方式构建嗜热蛋白质的特征向量。然后,对特征进行贡献度分析,挑选前179维特征重要性评估(Variable Importance Measures,VIM)得分最高的特征作为模型的最优特征子集。通过多种机器学习算法的比较,将MLP作为最终的分类算法,构建基于多特征融合的嗜热蛋白质类型预测模型。
1 嗜热蛋白质预测模型
在本研究中,首先嗜热蛋白质使用G-gap二肽组成、CTDC和MAAC三种特征提取方法进行表征,共构建了499维特征向量,然后使用VIM分析各个特征的贡献度以挑选最优特征子集,最后利用MLP来构建最终的预测模型。实验步骤如图1所示。

图1 基于FF和VIM的嗜热蛋白质预测流程
2 蛋白质序列特征表达模型
蛋白质序列是由20种氨基酸之间相互脱水缩合形成的。一条长度为N的[8]蛋白质P可以表达为:
P=R1R2…Ri…RN
式中,每一个R代表这条蛋白质中的一个氨基酸残基。例如R1代表蛋白质P的第一个氨基酸残基,R2代表蛋白质P的第二个氨基酸残基,Ri代表蛋白质P的第i个氨基酸残基,则RN就是第N个氨基酸残基。
2.1 多段氨基酸组成(MAAC)
MAAC是在氨基酸组成上的进一步演变,该方法通过计算组成序列氨基酸的频率来表征蛋白质序列[9],与传统氨基酸组成方法相比,MAAC联合蛋白质两端区域与中间段的氨基酸信息。本研究中将整段蛋白质序列分成三段,分别是N端氨基酸区域、中间段以及C端氨基酸区域。MAAC把蛋白质的空间位置信息融进特征中,一条蛋白质信息将由20维转变为60维。
SAAC=[S1,S2,S3]T
(2)
Sj=[a1j,a2j,a3j,…,a20j]T,(j=1,2,3)
(3)
(4)
式(2)中,S1代表前三分之一的蛋白质序列的氨基酸组成表征,S2代表中间三分之一的蛋白质序列的氨基酸组成表征,S3代表后三分之一的蛋白质序列的氨基酸组成表征。式(3)中aij代表第j段氨基酸序列的第i种氨基酸出现的频率[10]。式(4)中cij代表统计第j段氨基酸序列中第i种氨基酸的个数。
2.2 G-gap二肽组成
二肽组成方法通过计算蛋白质序列中任意两个相邻氨基酸组成的二肽的频率形成特征向量来表征蛋白质[11]。而20种基础氨基酸两两的排列组合得到的二肽形式共有400种,因此该方法将一条蛋白质序列表征为400维的数字特征向量:
D=[f1,f2,…,f400]T
(5)
(6)
式中,vi表示第i种相邻氨基酸对组成的二肽出现的次数,fi表示第i种相邻氨基酸对的频率。在传统的二肽组成方法的基础上进一步提出G-gap二肽组成[12]方法,其公式如下:
(7)
(8)

2.3 组成转变分布组成(CTDC)
CTDC特征提取方法将组成蛋白质的20种基础氨基酸[13]分成三类,以疏水性为例,所有的氨基酸都可以分为极性、中性和疏水性三种类别。然后,分别统计每一类氨基酸残基在整个蛋白质序列中出现的频率,记作n(x),计算每一个类别所占的百分比,公式如下:
(9)
式中,g1代表类别一极性[9],g2代表类别二中性,g3代表疏水性,其中N表示蛋白质氨基酸残基的总个数。
3 特征重要性评估(VIM)
本文使用MAAC、G-gap二肽组成和CTDC三种特征提取方法,共同构建向量维度为499维,使用VIM来挑选影响模型最大的特征子集,并以此缩减建模型输入变量的特征数。本文使用随机森林(Random Forest,RF)来进行特征重要性评估,使用Gini指数作为评价指标来衡量特征对于模型的重要度[14]。
输入特征为X1,X2,…,XM,其Gini指数的计算公式如下:
(10)
式中,K指数据的类别数,嗜热蛋白预测属于二分类问题,所以K=2。pmk表示节点m中类别k所占的比例。

(11)
式中,GIl与GIr为m节点分枝后的两个节点的基尼指数。若特征Xj在第i棵树中出现的频率为M,则特征Xj在第i棵树的贡献度为:
(12)
若RF中分类树的数量为n棵,则特征Xj的Gini指数为[15]:
(13)
4 多层感知机(MLP)
MLP最重要的一个特点是多层,除了输入输出层,它中间可以包含多个隐藏层。本文的MLP模型如图2所示,包含两个隐藏层,总共四层结构[16]。
从图2可知,MLP的层与层之间的所有神经元都有连接。MLP第i层是输入层,h层是隐藏层,最右边一列代表输出层。每一层输出都是上层输入的线性函数,无论MLP有多少层,输出都是输入的线性组合。而激活函数的使用,能够给神经元引入非线性功能,使得MLP可以任意逼近任何非线性函数,这样MLP就可以适用于更多的非线性模型中。本文模型激活函数使用的是Relu函数,表达式如下所示:
Relu(x)=max(x,0)
(14)

图2 MLP原理图
输入层为嗜热蛋白质序列样本,用向量X=(x1,x2…xn)表示。第一个隐藏层与输入层之间的解析表达式为:
(15)

(16)

输出层o为:
o={+1,-1}
(17)
式中的+1代表嗜热蛋白质,而-1代表常温蛋白质。
MLP的学习过程就是根据训练集来对神经元之间的连接权以及每个功能神经元的阈值进行调整。
5 实验结果与分析
5.1 数据集来源
本文研究的嗜热蛋白质和常温蛋白质样本数据集来源于文献[2],从UniProt中根据最适宜温度的标记选取嗜热蛋白质和常温蛋白质序列,对于选取的蛋白质序列,包含“B”“J”“O”“U”“X”和“Z”的序列被剔除,并使用CD-HIT软件去除冗余,同时序列相似度在40%以上也被删除。通过以上步骤,获得915条嗜热蛋白质序列以及793条常温蛋白质序列,最终以共1708条蛋白质序列作为本文的数据集N,其表示如下:
N=NT∪NM
(18)
式中,NT表示915条嗜热蛋白质序列的数据集,NM表示793条常温蛋白质序列的数据集。
5.2 交叉验证与性能评价
分类预测常用的交叉验证方法有K折交叉验证和留一法。K折交叉验证方法基本原理是将样本数据有n个的集合S分割为k个大小相同的互斥子集,其表达式为:
S=s1∪s2∪s3…∪sk
(19)
k为子集个数,然后依次从k个子集中选出1个子集作为验证集,其余k-1个子集作为测试集,进行k次模型训练和验证。而留一法是将样本分割成n份,即一个子集包含一个样本,每次选出一个样本作为验证集,其余n-1个样本作为训练集,进行n次模型训练和验证。
对比其余交叉验证方法,留一法[18]被认为是最严格最客观的,所以本文主要运用留一法进行交叉验证。
针对嗜热蛋白质分类问题,通过把样本的真实类别与模型的预测类别进行对比,可以形成真正例、假正例、真反例、假反例四种情况,如表1所示[19]。

表1 混淆矩阵表
常用的模型性能评价指标主要包括:准确率(Acc)、马修斯相关系数(MCC)、查准率(P)、敏感性(Sn)和特异性(Sp),具体如下。
准确率(Acc)定义为:
(20)
敏感性(Sn)定义为:
(21)
特异性(Sp)定义为:
(22)
查准率(P)定义为:
(23)
马修斯相关系数(MCC)定义为:
MCC=
(24)
本文利用以上性能评价指标对预测结果进行分析。
5.3 实验结果分析
将嗜热蛋白质看作正样本,将常温蛋白质看作负样本。本文实验数据集存在正负样本不平衡的状态,正负样本不平衡可能会影响最后的预测结果,降低模型的泛化性能。为此,本实验对于样本采取欠采样方法随机删除部分正样本,使得正样本个数与负样本数据基本持平,确保实验结果的精确性与可靠性。本文针对现有模型特征工程表征能力不足的问题,提出使用MAAC、G-gap二肽组成和CTDC三种蛋白质提取方法分别对嗜热蛋白质进行表征,然后将提取的三种特征进行特征融合构建样本的特征,MAAC构建的蛋白质特征向量为60维,CTDC用39维特征表征蛋白质,G-gap二肽使用400维特征,共有499维特征向量,特征融合虽然可以使得蛋白质信息表达得更加全面,但是也带来了由于维度过大增加计算复杂度的问题,同时还会存在信息冗余现象,为了解决这个问题并减少训练时间,本实验对特征进行了特征重要度分析,得出每个特征的VIM得分。
5.3.1 嗜热蛋白质特征变量重要性分析
本文使用VIM分析各个特征的贡献度。RF是一种常见的机器学习算法,其在度量特征重要性方面相比于其他机器学习算法更具优势,所以本文使用包含250棵决策树的RF模型,获得了嗜热蛋白质特征VIM得分,如图3所示。

图3 嗜热蛋白质特征重要性分析
从图3可知,特征排序是按照VIM得分由高到低依次排列的。为了更清晰明确地得出对于预测贡献度较大的特征,我们选取VIM得分较高的前20维特征,如图4所示。VIM得分越高说明特征贡献度越高,从图4可知在贡献度较高的前20维特征中,CTDC的贡献率是最高的,前六维的特征均来自CTDC,总贡献率大约是占据第二位的多段氨基酸的贡献度的三倍,说明CTDC与嗜热蛋白质预测精度的提升有很大的关联。
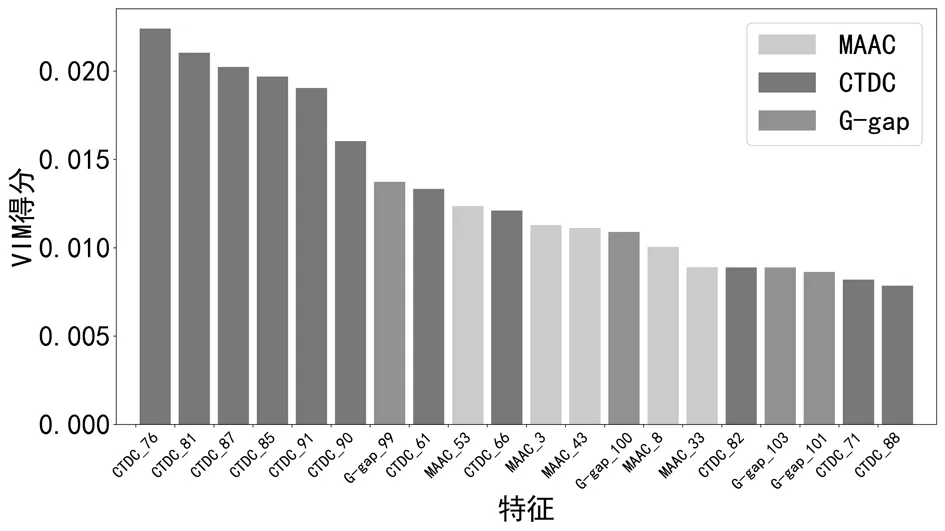
图4 特征重要性较高的前20维特征
5.3.2 嗜热蛋白质特征筛选
通过VIM算法,对特征重要度进行分析,按VIM得分从高到低对特征进行排列。特征的排序越靠前,其对嗜热蛋白质的预测就越重要。基于特征排序,以设定VIM得分阈值来挑选最优特征子集,通过计算不同VIM得分阈值组成的特征子集所建模型的精确度来确定最优阈值。其实验结果如图5所示,当VIM得分设定在0.0013时,即所选特征子集包含了VIM得分在0.0013以上的特征,精确度最高达到93.19%。VIM得分在0.0013以上的共179维,则对应的特征子集由得分最高的前179维特征组成,即为嗜热蛋白质的最优特征子集。多层感知机的各项参数为:solver设置为lbfgs, alpha设置为1e-2,隐藏层设置为2层,每层78个神经元,随机种子设置为100。

图5 特征VIM得分精度图
5.3.3 不同特征提取方法的比较
为了更加客观地评价本文所建融合特征模型的预测性能,使用MAAC、G-gap二肽组成和CTDC三种特征提取方法进行特征融合来表征嗜热蛋白质,然后提取嗜热蛋白质的最优特征子集来构建嗜热蛋白质的最终融合特征,与使用G-gap、CTDC、MAAC、MAAC+CTDC、MAAC+G-gap、CTDC+G-gap等6种蛋白质特征提取方法所构建的嗜热蛋白质预测模型分别进行对比,并使用留一法进行交叉验证,结合多种评价指标进行对比,对比结果如表2所示。由表2可知,基于本文特征构建的预测模型精确度高达93.19%,高于其余6个模型的预测精度,且在查准率(P)、敏感度(Sn)、特异性(Sp)、马修斯相关系数(MCC)这四个指标上也高于其余6个模型。且本文特征所建模型的敏感度(Sn)为93.69%,特异性(Sp)为92.69%,说明基于本文特征所构建的模型对于正负样本的预测效果都很好,几乎达到了平衡。进一步说明本文使用的特征融合预测模型在提高嗜热蛋白质的预测精度上有很大的作用。
5.3.4 不同分类算法在数据集上的精确度对比
本文采用的特征融合表征方法在一定程度上提高了预测精度,为了更客观地评价多层感知机构建模型的预测性能,将MLP与线性判别分析 (Linear Discriminant Analysis,LDA)、朴素贝叶斯(Nave Bayes,NB)、决策树 (Decision Tree,DT)、K近邻 (k-Nearest Neighbors,KNN)4种分类算法所构建的模型进行对比,为了保证一致性,实验的数据集均使用筛选的179维最优特征子集,并采用留一法进行交叉验证,对比结果如表3所示。
从表3可知,DT构建的模型预测性能最低,在精确度(Acc)、查准率(P)、敏感度(Sn)、特异性(Sp)、马修斯相关系数(Mcc)五项评价指标当中最低,而LDA在五项评价指标中表现略低于MLP,但均高于其余三种分类算法。NB的整体预测性能比DT略胜一筹,但是比KNN的预测性能稍低。MLP的性能最佳,预测精度达到了93.19%,比其余四种对比算法分别提高了1.14%、4.29%、7.51%、3.22%。在敏感度、特异性两个评估指标上高达92.6%以上,证明使用MLP能构建更优的预测模型。

表2 不同特征预测结果对比

表3 不同分类算法对分类效果的影响
为了更加直观地比较MLP、LDA、KNN、DT、NB这五种分类算法的分类效果,绘制了这五种分类算法的P-R曲线图与ROC曲线图,如图6、图7、图8所示。从图6可以发现,多层感知机MLP的P-R曲线图包裹住了其余四种分类算法的曲线,证明其分类效果更好;同时从图7可得,MLP的曲线图也包住了其余分类算法的曲线,而ROC曲线的面积越大,分类性能更好;从图8我们可以看出MLP的ROC面积达到了0.98。从以上三个指标综合分析可得,MLP在嗜热蛋白质的分类性能上超越了其余的分类算法,分类效果更佳。
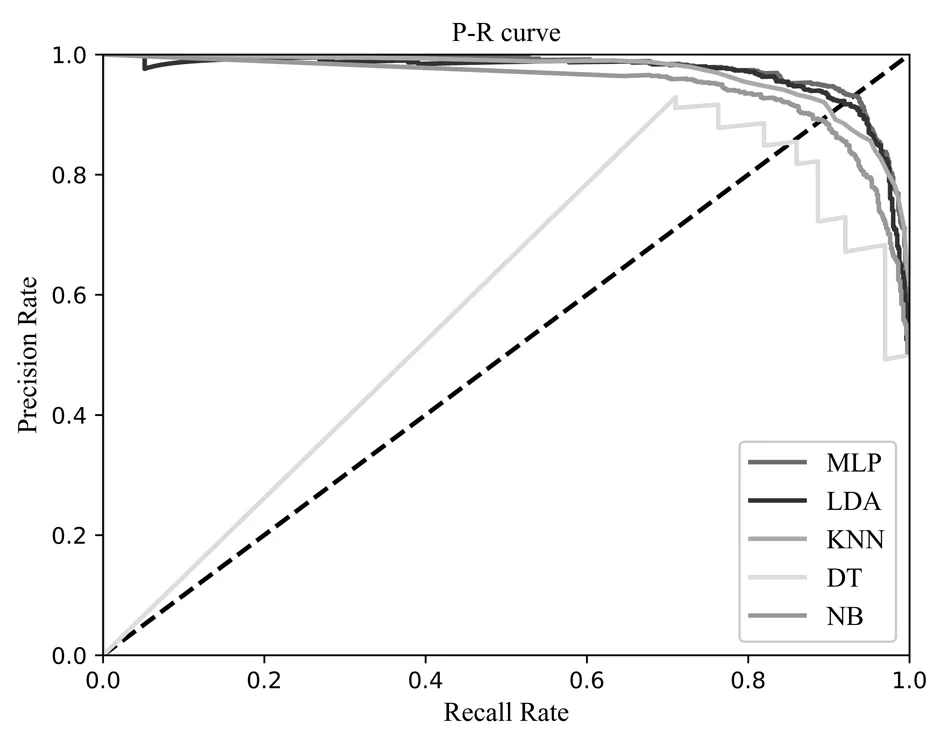
图6 不同分类算法的P-R曲线图
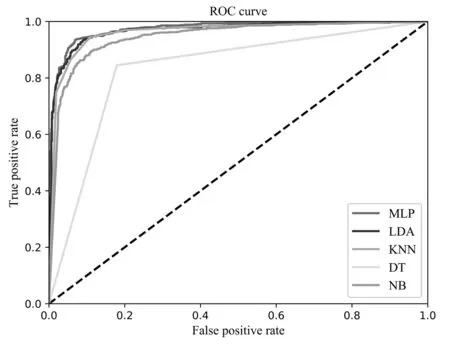
图7 不同分类算法的ROC曲线图

图8 多层感知机的ROC曲线图
5.3.5 与现有模型对比
为了验证模型的有效性,与使用同一数据集的其余实验进行对比是很有必要的,所以我们同文献[2]和文献[20]的实验进行对比,结果如表4所示。本文方法的特异性指标Spe为0.9269,高于文献[2]的方法而低于文献[20]的方法;但精确度指标Acc为0.9319,敏感性指标Sen达到0.9369,均高于其余两种方法。结合以上分析可知,本文方法对于嗜热蛋白质预测性能的提升是有效的。

表4 同一数据集上不同方法的预测结果对比
6 总结
本文针对单一特征表征蛋白质信息不充分的问题,提出了基于特征融合的概念,将MAAC、G-gap二肽组成和CTDC三种蛋白质特征提取方法结合起来用于表征嗜热蛋白质的特征向量,利用VIM方法筛选最优特征子集,特征维度降低的同时减少冗余信息与降低时间复杂度。经过实验对比分析发现,本文所提出的融合特征表达的预测性能更优,选用MLP作为分类算法,并采用留一法在多个分类算法上进行交叉验证,精度高达93.19%。结果表明将多个特征融合起来,并使用MLP来构建预测模型对于嗜热蛋白质预测性能的提高是有效的。
猜你喜欢
肝博士(2022年3期)2022-06-30
安庆师范大学学报(自然科学版)(2021年1期)2021-11-28
海外星云(2021年9期)2021-10-14
食品安全导刊(2021年21期)2021-08-30
湖南饲料(2021年3期)2021-07-28
兽医导刊(2020年13期)2020-12-31
合肥工业大学学报(自然科学版)(2020年2期)2020-03-23
南京大学学报(数学半年刊)(2020年1期)2020-03-19
医学研究杂志(2015年9期)2015-07-01
都市丽人(2015年4期)2015-03-20
