
伴有PAXIP1和NOS3基因突变的额颞叶痴呆1例
2021-09-25 02:17赵修琦南善姬
中国实验诊断学 2021年9期
王 瑞,张 雷,赵修琦,南善姬
(吉林大学第二医院 神经内科,吉林 长春130041)
额颞叶痴呆(FTD)是一组以进行性额颞叶变性(FTLD)为共同病理特征的临床综合征,发病率每年约为1.6-4.1/10万,性别无差异。在65岁以下人群中,FTD是仅次于阿尔茨海默病的第二常见痴呆。FTD是一种异质性疾病,存在不同的临床表型及神经病理类型,临床上往往以明显的人格、行为改变和语言障碍为特征,可以合并运动神经元病[1]。本文报道1例伴有PAXIP1(PTIP)基因突变(E204K)和NOS3(nitric oxide synthase 3)基因突变(1-4号外显子区域重复变异)额颞叶痴呆病例。
1 临床资料
患者,男,63岁,因性格改变,记忆力减退2年,加重半年入院。2年前患者妻子、女儿发现患者易怒、固执,伴有近记忆下降,不记得自己刚刚说过的话、做过的事。半年前患者出现偏执、淡漠,语言减少、用词单一,经常答非所问、找词困难,对家人缺乏关心。患者不认识熟悉的老邻居,不能独立完成烹饪。既往“高血压病”病史40年;“头外伤”病史4年,饮酒史30余年。入院查体:血压139/92 mmHg,高级认知功能下降,余神经系统查体正常。血常规、肝肾功、离子、血糖、血脂、甲功五项、免疫常规等化验无异常。头部CT和MRI左侧颞叶脑回变窄、脑沟加深,左侧脑室颞角扩张明显,右颞极可见脑沟加深。提示双侧颞叶萎缩,左侧重于右侧。18F-FDG头部PET/CT断层显像提示左侧额叶、顶颞叶、后扣带回、尾状核及右侧壳核糖代谢明显减低,右侧额叶、后扣带回、尾状核及左侧壳核糖代谢减低,中脑、脑桥、右侧顶叶、感觉运动区、颞叶、左侧颞中回及双侧小脑糖代谢增高。11C-PIB头部PET/CT断层显像提示皮层各叶放射性分布尚均匀,未见异常放射性滞留,考虑为PIB显像阴性。见图1。
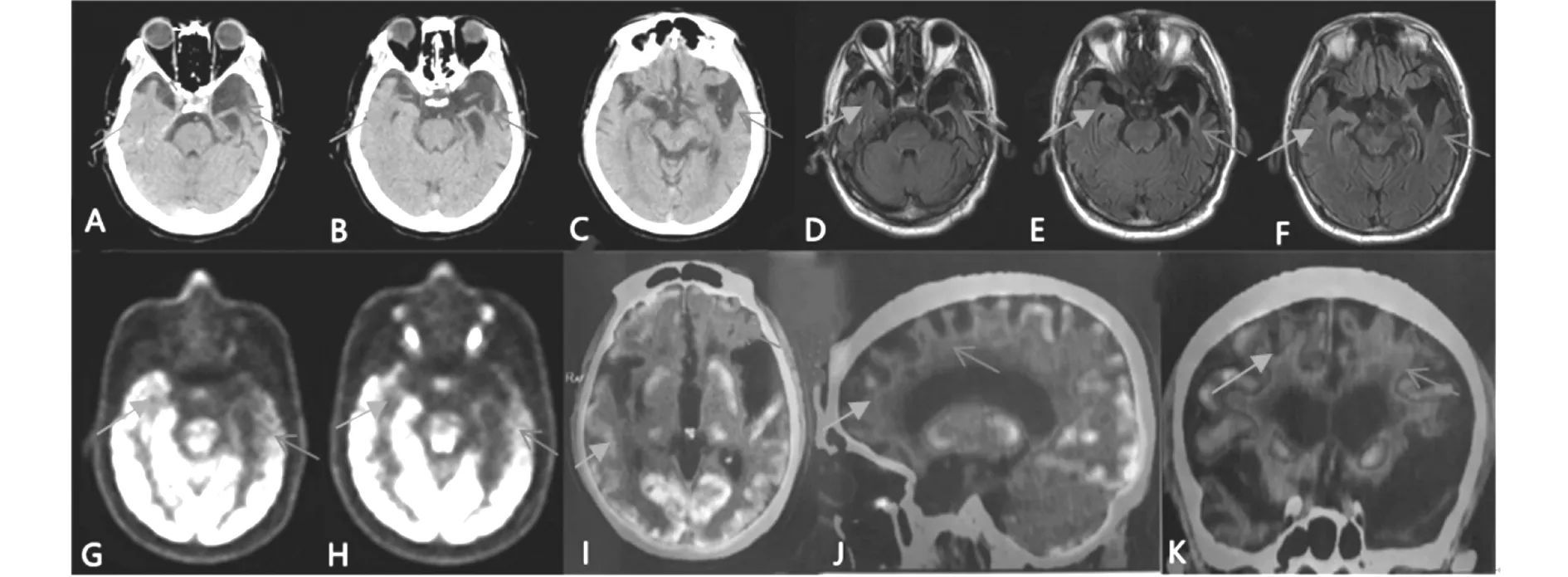
图1 患者影像学
神经心理量表检查:简易智能精神量表(MMSE)24分(定向力7分;回忆能力0分;语言能力7分;书写能力0分);蒙特利尔认知评估量表(MoCA)14分(视空间与执行功能5分;语言重复0分;语义流畅性0分;抽象0分;延迟回忆0分;定向力4分);听觉词语学习测验(AVLT)总分8分;波士顿命名测验13分;顺向数字广度6分,逆向数字广度4分;汉密尔顿焦虑量表7分,汉密尔顿抑郁量表4分;NPI(神经精神问卷)32分。
经患者和家属同意,采集患者抗凝血5 ml,进行全外显子基因检测(北京海思特医学检验实验室)。基因检测结果:①PAXIP1基因存在杂合变异,c.610G>A(鸟嘌呤>腺嘌呤),导致氨基酸改变p.E204K(谷氨酸>赖氨酸)。见图2。HGMDpro数据库报道情况:变异位点c.610G>A未见报道。根据ACMG指南,该变异可评级为临床意义未明变异(PM2:正常人群变异数据库未见报道)。SIFT、Polyphen-2和Mutation Taster软件预测结果分别为Tolerated、Benign、Tolerated。提示该突变对蛋白质功能的影响比较小或者没有影响。②NOS3基因1-4号外显子区域存在重复变异。其中,1、3、4号外显子的重复变异已经qPCR检测证实。HGMDpro数据库报道情况:该基因1-4号外显子重复变异未见报道。根据ACMG指南,该变异可评级为临床意义未明变异。若此变异为致病性变异,理论上有可能增加相应疾病的患病风险。
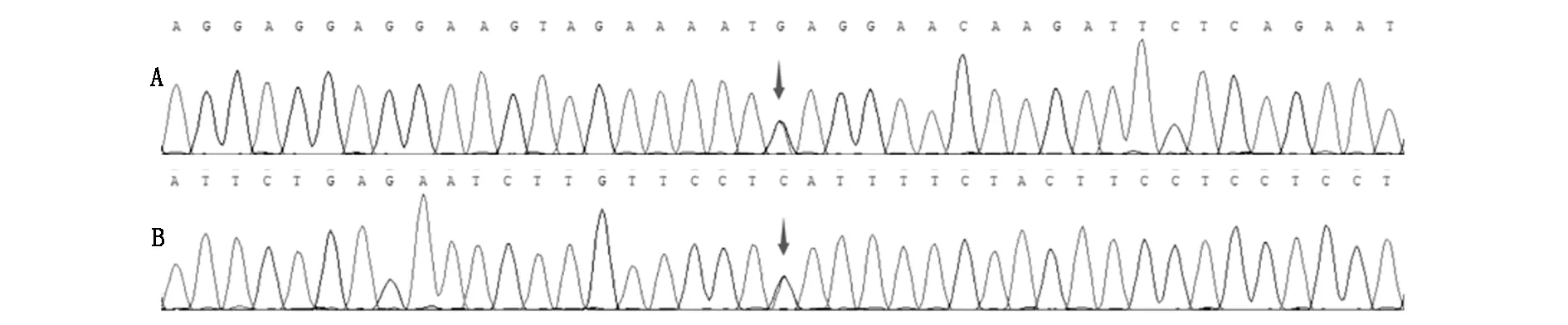
图2 测序结果
2 讨论
FTD最早由捷克神经病学家Pick于1892年描述。1911年,Alzheimer对Pick的病例进行了组织学分析,发现其神经元中有嗜银细胞质包涵体,将此病命名为“Pick病”。20世纪70年代后,Mesulam、Snowden等人相继提出了原发性进行性失语、语义性痴呆的概念。1994年,FTD的第一个诊断标准由英国和瑞典的两个研究小组合作制定。根据早期症状,FTD可分为三种亚型:行为变异性额颞叶痴呆(bvFTD),最常见,约占所有FTLD的56%,表现为早期行为和执行功能障碍,以额叶萎缩为主。进行性非流利性失语(PNFA),此型约占FTLD的25%,表现为语言输出障碍和语法缺陷,以左侧额叶和岛叶萎缩为主。语义性痴呆(SD),发病年龄稍晚,进展相对较慢,表现为进行性语义和命名方面的障碍,特征是失去词语的意义和概念,左侧颞叶前部较对侧萎缩严重。SD也可以从右侧起病,出现早期情绪障碍和对熟悉面孔的失认。随着病情进展,患者会出现全面的认知障碍和运动障碍,包括帕金森综合征。此外,一些FTD合并存在运动神经元疾病。
本例患者以性格改变和记忆力减退为首发症状,症状进行性加重,出现偏执、淡漠、言语减少、找词困难、面孔失认。神经心理量表检查提示患者执行、语言、抽象、记忆及精神情感障碍。神经影像学上可见左侧颞叶萎缩,脑室颞角明显扩张,右侧颞叶颞极萎缩。18F-FDG头部PET/CT断层显像提示大脑前部糖代谢减低,左侧较右侧明显。11C-PIB头部PET/CT断层显像未见异常放射性滞留,PIB显像阴性。综上,该例患者符合FTD的SD亚型。
PAXIP1基因位于第7号染色体,编码1个核蛋白,具有6个BRCT(breast cancer carboxy-terminal))结构域,在DNA修复通路上发挥作用[2]。2005年,Rosa Rademakers 报道在一个荷兰早发性AD家系中发现PAXIP1基因10号外显子存在一个同义突变(g.38030G>C)。该基因是否为致病基因或在发病过程中是否发挥作用尚不清楚[3]。2015年,Jason A Chen等运用人类外显子芯片技术对224例AD、168例FTD、48例PSP和224例正常对照进行了一项多中心研究。他们在AD病例中检测到PAXIP1基因存在5种突变(1个同义突变,4个错义突变)。脑区PAXIP1基因表达情况研究中发现AD病例前额皮层、视觉皮层和小脑区域PAXIP1表达升高[4]。这些研究结果似乎都支持PAXIP1与AD发病有关,但仍缺乏直接证据。本病例全外显子基因检测发现PAXIP1基因存在杂合变异(E204K),经SIFT、Polyphen-2和Mutation Taster软件预测结果分别为Tolerated、Benign、Tolerated,提示该突变对蛋白质功能的影响比较小或者没有影响。此外,本病例NOS3基因1-4号外显子区域存在重复变异。NOS3基因与晚发性AD易感相关,报道为常染色体显性遗传,理论上有一条染色体发生致病性变异即可增加相应疾病的患病风险。目前为止,该基因1-4号外显子重复变异未见报道(https://omim.org)。
综上,本文报道的病例临床和功能影像学均符合额颞叶痴呆的诊断,存在PAXIP1基因和NOS3基因突变。虽然,这两种基因都与AD的发病有关,但是PAXIP1基因突变(E204K)和NOS3基因突变(1-4号外显子区域重复变异)尚不能明确其临床意义。这两种突变是否参与到FTD的发病机制中有待更多病例的报道和进一步研究。
猜你喜欢
保健与生活(2021年21期)2021-11-11
科技资讯(2020年32期)2020-12-28
上海针灸杂志(2020年1期)2020-02-11
家庭科学·新健康(2019年9期)2019-10-21
医学信息(2018年5期)2018-04-20
意林·少年版(2018年24期)2018-01-05
公务员文萃(2017年7期)2017-07-21
饮食科学(2017年4期)2017-05-03
读者(2016年18期)2016-08-23
医学研究杂志(2015年9期)2015-07-01
