
苦参碱固体脂质纳米粒的制备及其体外透皮研究
2021-09-23 09:10付丽娜李伟泽韩文霞杨黎彬
中草药 2021年18期
付丽娜,赵 宁,李伟泽,韩文霞,李 健,杨 婷,杨黎彬
西安医学院药学院,陕西 西安 710021
苦参碱(matrine)是来源于豆科槐属植物苦参Sophora flavescensAir.的一种生物碱类天然药物, 具有抗炎、抗菌、抗病毒、抗肝纤维化、抗肿瘤、抗心律失常、抑制中枢神经和调节免疫力等药理作用[1-7]。目前在临床上广泛应用的苦参碱制剂主要有注射剂、胶囊剂、片剂和栓剂等[8]。但是,传统苦参碱制剂普遍存在半衰期短、需多次给药,易造成肝肾聚集中毒等问题;并且由于苦参碱的苦味强烈持久,患者用药依从性差[9-10]。这些限制了苦参碱口服制剂与注射制剂的广泛应用,因此,开发苦参碱的外用缓释给药系统具有重要意义。固体脂质纳米粒(solid lipid nanoparticles,SLN)是粒径为纳米级别的一种新型实体骨架的微粒给药系统,可作为大分子、小分子药物的载体,具有良好的促进药物透皮吸收作用和皮肤贮库效应,能够增加药物的稳定性,可提高药物对皮肤黏膜的穿透性而提高其生物利用度,且药物泄露问题较少,可通过透皮、口服、注射与肺部等多途径给药,同时其成本低与利于大规模的生产,是微乳、脂质体、聚合物纳米粒等的替代品[11-13],因此SLN具有良好的应用前景。据此,本实验研究了以微乳-低温固化法制备苦参碱固体脂质纳米粒(matrine solid lipid nanoparticles,MA-SLN)的成型工艺及其相关药剂学性能,并通过体外透皮给药考察了MA-SLN透皮给药的释药行为,从而为苦参碱提供了一种基于纳米微粒的新型外用缓释给药系统。
1 仪器与材料
1.1 主要仪器
Agilent1260高效液相色谱仪,美国安捷伦科技有限公司;Verios 460扫描电子显微镜(SEM),美国FEI公司;JEM-2100F透射电子显微镜(TEM),日本电子株式会社;X’Pert PRO X-射线衍射仪(XRD),荷兰帕纳科公司;ZEN-3600激光粒度仪,英国马尔文公司;Quintix224-1 CN Sartorius分析天平,赛多斯科学仪器北京有限公司;Stare系统DSC差式扫描量热仪(DSC),瑞士梅特勒-托利多公司;Allegra 64R Centrifuge离心机,美国Beckman公司。
1.2 药材与试剂
苦参碱对照品,中国食品药品检定研究院,质量分数110805-201803;苦参碱,质量分数≥98%,批号XC20190410,西安小草植物科技有限责任公司;大豆卵磷脂,上海太伟药业有限公司;硫酸鱼精蛋白,质量分数99%,Sigma公司;单硬脂酸甘油酯,南昌华鑫医药化工有限公司;普朗尼克F-68,天津市科密欧化学试剂有限公司;其他试剂均为分析纯,水为双蒸水。
1.3 动物
健康昆明种小鼠,雌雄各半,体质量(20±2)g,批号20191003,购自西安交通大学医学部。所有动物实验遵循陕西省有关实验动物管理和使用的规定,均符合3R原则。
2 方法与结果
2.1 MA-SLN的制备
采用微乳-低温固化法[14-15]制备MA-SLN。称取苦参碱50 mg,适量单硬脂酸甘油脂和大豆卵磷脂,并加入5 mL无水乙醇作为油相;另取0.2 g普朗尼克F-68,加入一定量纯化水作为水相;油相与水相分别置于65 ℃水浴中,使溶解;并于一定转速条件下将水相缓慢加入油相,搅拌乳化,再高速匀质4 min后将乳化体系分散到冷水中,300 r/min条件下搅拌固化20 min,过0.45 µm微孔滤膜,即得MA-SLN。同法不加苦参碱,制备空白脂质纳米粒(blank solid lipid nanoparticles,B-SLN)。
2.2 苦参碱检测方法的建立
2.2.1 供试品溶液的制备 取一定量MA-SLN于2 mL的离心管中,加入同体积10 μg/mL鱼精蛋白溶液,静置3 min,于10 ℃、13 000 r/min条件下离心60 min,取上清液,过0.22 μm滤膜,即得。
2.2.2 色谱条件 色谱柱为Agilent Zorbax SB-C18(150 mm×4.6 mm,5 µm);流动相为甲醇-水(85∶15);体积流量1.0 mL/min;柱温35 ℃;进样量为20 µL;检测波长220 nm。
2.2.3 线性关系考察 精密称取苦参碱对照品,加蒸馏水配制成质量浓度分别为20、30、60、100、200、400 µg/mL的对照品溶液,分别标为1~6号,取上述各对照品溶液过0.22 µm微孔滤膜,取滤液20 μL用HPLC按照“2.2.2”项下色谱条件测定,以峰面积为纵坐标(Y)、质量浓度为横坐标(X),进行线性回归,计算得回归方程为Y=6.649 2X+100.99,r2=0.999 8,表明苦参碱在20~400 µg/mL时质量浓度与峰面积线性关系良好。
2.2.4 专属性考察 取苦参碱对照品溶液,B-SLN和MA-SLN,其中B-SLN和MA-SLN按照2.2.1项下方法处理,并按“2.2.2”项下色谱条件检测,结果见图1。实验结果显示,该色谱条件下,色谱峰分离度好,同时峰形稳定,无干扰。

图1 苦参碱对照品 (A)、B-SLN (B) 和MA-SLN (C) 的HPLC图Fig.1 HPLC of matrine reference substance (A),B-SLN (B) and MA-SLN (C)
2.2.5 精密度试验 取1、3、6号对照品溶液,按“2.2.2”项色谱条件测定峰面积,每天0、2、4、6、8、10 h进样,连续进样3 d,测定峰面积,考察日内和日间精密度,结果其日间和日内精密度RSD分别为1.63%和1.87%。
2.2.6 重复性试验 精密量取同一批次MA-SLN 6份,按照“2.2.1”项下处理后,按“2.2.2”项下色谱条件进样测定峰面积,计算苦参碱质量浓度,RSD值为1.76%,表明该方法重复性良好。
2.2.7 稳定性试验 将同一批次供试品溶液室温下放置,分别于0、3、6、12、24、48 h取20 μL注入HPLC测定,苦参碱峰面积的RSD为0.92%,表明供试品溶液在48 h内稳定。
2.2.8 加样回收率试验 取9份B-SLN 0.5 mL,置于2 mL离心管内,分别加入80、100、120 μg/mL 苦参碱对照品溶液0.5 mL,再按2.2.1项下方法加10 μg/mL鱼精蛋白溶液1 mL,静置3 min,于10 ℃、13 000 r/min条件下离心60 min,取上清液,过0.22 μm微孔滤膜,按照“2.2.2”项下色谱条件测定,测得平均加样回收率为99.77%,RSD为1.87%。
2.3 MA-SLN处方筛选
2.3.1 MA-SLN包封率测定 采用鱼精蛋白凝聚法测定包封率[16],精密吸取MA-SLN样品0.8 mL,置于2 mL离心管,加10 μg/mL鱼精蛋白溶液0.8 mL,混匀,静置3 min,于10 ℃、13 000 r/min条件下离心60 min,取上清液1 mL置于10 mL量瓶中,定容,过0.22 μm微孔滤膜,取20 μL按照“2.2.2”项下色谱条件测定苦参碱的量,计算包封率。
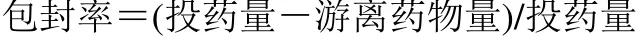
2.3.2 粒径和ζ电位的测定 取MA-SLN用蒸馏水稀释至10倍,使溶液呈现半透明乳光色,用激光粒度仪测定其粒径与ζ电位。
2.3.3 评价指标的确定 包封率、粒径和ζ电位为SLN质量控制的重要指标。因此,本实验以包封率、粒径和ζ电位为MA-SLN处方筛选的评价指标。其中,包封率是纳米粒质量控制的一个重要的因素,也是提高药物治疗效果,减少药物剂量降低不良反应的关键因素。因此,设定包封率在0~100%内,每增大10%,加1分,满分10分,占比60%;SLN的粒径一般为50~1000 nm,粒径越小,纳米粒的透皮吸收效果越好,根据实验过程中不同工艺所测MA-SLN的粒径范围,设定以490 nm为基数,减小40 nm,加1分,满分10分,占比20%;ζ电位在一定程度上反应粒子的荷电情况,对维持体系的稳定性有很大影响,当微粒表面电位的绝对值大于30 mV时,微粒间由于存在较大的静电斥力而有利于制剂稳定性的提高并使粒径保持均匀,因本实验所制备的MA-SLN带负电荷,因此设定,ζ电位在-60~0 mV,比0 mV小6 mV,加1分,满分10分,占比20%。将评分进行综合加权处理,并以处理后的综合得分为评价指标。
2.3.4 正交试验设计与结果 根据前期单因素中各因素对实验结果的影响,选择单硬脂酸甘油酯(A)、卵磷脂(B)的用量和油水两相比例(C)进行处方优化考察,以综合得分为评价指标,采用L9(34)表进行正交设计实验,正交试验的因素水平、实验设计与结果见表1,方差分析见表2。由表1、2可知,各因素对MA-SLN包封率和粒径的综合的影响大小依次为A>B>C,单硬脂酸甘油酯(A)和卵磷脂(B)对MA-SLN包封率和粒径有显著影响,根据正交表直观分析,选择最佳工艺条件为A2B3C2,即MA-SLN的最佳制备处方为苦参碱50 mg、单硬脂酸甘油酯0.15 g、大豆磷脂0.3 g、普朗尼克F-68 0.2 g、油相5 mL和水相体积10 mL,制得的MA-SLN的包封率较高、粒径小、ζ电位合适。
表1 L9(34)正交试验设计及结果 ( ±s,n = 3)Table 1 Design and results of L9(34) orthogonal test ( ±s,n = 3)

表1 L9(34)正交试验设计及结果 ( ±s,n = 3)Table 1 Design and results of L9(34) orthogonal test ( ±s,n = 3)
试验号 A/g B/g C D (误差) 包封率/% 包封率得分 粒径/nm 粒径得分 ζ电位/mV ζ电位得分 综合得分1 0.10 (1) 0.10 (1) 1∶1 (1) (1) 52.07±1.36 5.00 361.23±8.49 4.00 -29.97±1.87 5.33 4.87 2 0.10 (1) 0.20 (2) 1∶2 (2) (2) 52.40±1.23 5.00 210.23±6.92 7.67 -39.17±1.17 7.00 5.93 3 0.10 (1) 0.30 (3) 1∶3 (3) (3) 51.93±1.42 5.00 219.70±9.83 7.33 -40.83±1.83 7.33 5.93 4 0.15 (2) 0.10 (1) 1∶2 (2) (3) 58.60±4.07 5.33 207.97±9.85 7.67 -37.67±1.12 7.00 6.13 5 0.15 (2) 0.20 (2) 1∶3 (3) (1) 58.70±3.58 5.67 198.33±8.85 8.00 -39.60±2.29 7.33 6.47 6 0.15 (2) 0.30 (3) 1∶1 (1) (2) 58.50±3.12 5.67 193.67±7.09 8.00 -38.73±1.65 7.00 6.40 7 0.20 (3) 0.10 (1) 1∶3 (3) (2) 43.67±2.43 4.00 319.87±7.01 5.00 -30.77±1.69 5.67 4.53 8 0.20 (3) 0.20 (2) 1∶1 (1) (3) 49.87±2.12 4.67 256.63±8.95 6.33 -33.20±1.90 6.00 5.22 9 0.20 (3) 0.30 (3) 1∶2 (2) (1) 59.23±2.21 5.67 234.43±9.40 7.00 -36.46±1.78 6.33 6.07 K1 16.73 15.53 16.49 17.41 K2 19.00 17.62 18.13 16.86 K3 15.82 18.40 16.93 17.28 R 3.18 2.87 1.64 0.55 优选方案 A2 B3 C2

表2 方差分析Table 2 Analysis of variance
2.4 MA-SLN制备工艺优化
工艺优化的评价指标和评判方法同“2.3.3”项。研究表明,乳化体系的形成过程中需要充分的乳化速度和乳化时间才能保证乳化剂的效用[17]。因此,本实验用单因素实验法考察了不同搅拌时间、搅拌速度与不同稀释相体积对MA-SLN包封率、粒径和ζ电位的影响,按照“2.3.3”项将实验结果进行加权处理得出综合评分,最后根据综合评分筛选出最优成型工艺。
2.4.1 不同搅拌速度对MA-SLN包封率、粒径、ζ电位的影响 按照“2.3.4”项下确定的最优处方,在搅拌时间30 min,分散相体积30 mL的条件下,考察不同搅拌速度的MA-SLN综合评分的影响。结果见表3。
2.4.2 不同搅拌时间对MA-SLN包封率、粒径、ζ电位的影响 按照“2.3.4”项下确定的最优处方,在搅拌速度1000 r/min,分散相体积30 mL的条件下,考察不同搅拌时间的MA-SLN的综合评分的影响。结果见表4。
表3 搅拌速度对MA-SLN综合评分的影响 ( ±s,n = 3)Table 3 Effect of stirring speed on MA-SLN comprehensive score ( ±s,n = 3)

表3 搅拌速度对MA-SLN综合评分的影响 ( ±s,n = 3)Table 3 Effect of stirring speed on MA-SLN comprehensive score ( ±s,n = 3)
搅拌速度/(r·min-1) 包封率/% 包封率评分 粒径/nm 粒径评分 ζ电位/mV ζ电位评分 综合评分 600 43.77±1.30 4.00 387.70±5.22 3.00 -18.07±1.42 3.33 3.67 800 51.67±0.96 5.00 361.23±7.49 4.00 -29.33±0.93 5.33 4.87 1000 56.77±3.65 5.33 215.47±5.76 7.33 -41.20±1.28 7.33 6.13 1200 53.83±1.27 5.00 208.10±4.83 7.67 -42.40±0.95 7.67 6.07 1400 51.83±1.65 5.00 211.60±6.74 7.67 -38.17±1.56 7.00 5.93
2.4.3 不同分散相体积对MA-SLN包封率、粒径、ζ电位的影响 按照“2.3.4”项下确定的最优处方,在搅拌速度1000 r/min,搅拌时间30 min的条件下,考察不同分散相体积的MA-SLN的综合评分的影响。结果见表5。
表4 搅拌时间对MA-SLN综合评分的影响 ( ±s,n = 3)Table 4 Effect of stirring time on MA-SLN comprehensive score ( ±s,n = 3)
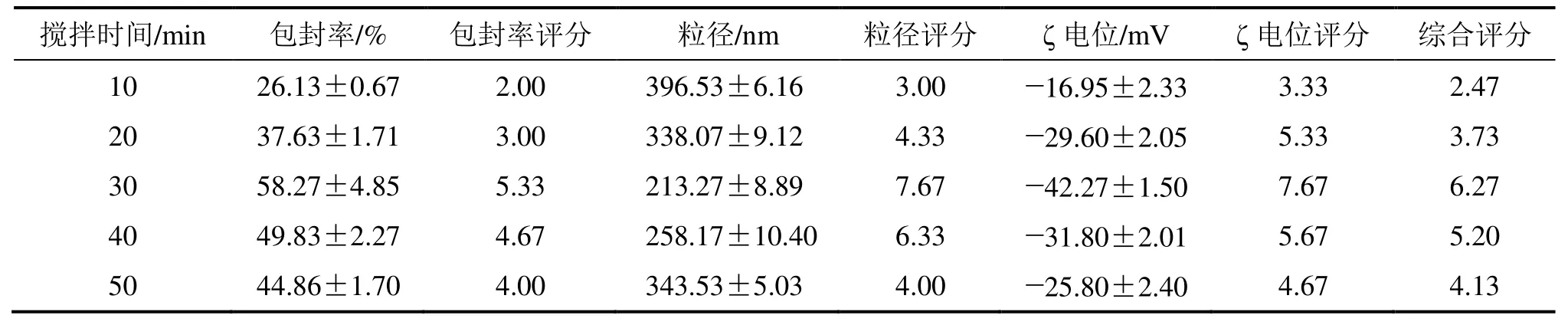
表4 搅拌时间对MA-SLN综合评分的影响 ( ±s,n = 3)Table 4 Effect of stirring time on MA-SLN comprehensive score ( ±s,n = 3)
搅拌时间/min 包封率/% 包封率评分 粒径/nm 粒径评分 ζ电位/mV ζ电位评分 综合评分 10 26.13±0.67 2.00 396.53±6.16 3.00 -16.95±2.33 3.33 2.47 20 37.63±1.71 3.00 338.07±9.12 4.33 -29.60±2.05 5.33 3.73 30 58.27±4.85 5.33 213.27±8.89 7.67 -42.27±1.50 7.67 6.27 40 49.83±2.27 4.67 258.17±10.40 6.33 -31.80±2.01 5.67 5.20 50 44.86±1.70 4.00 343.53±5.03 4.00 -25.80±2.40 4.67 4.13
表5 分散相体积对MA-SLN综合评分的影响 ( ±s,n = 3)Table 5 Effect of dispersed phase volume on MA-SLN comprehensive score ( ±s,n = 3)

表5 分散相体积对MA-SLN综合评分的影响 ( ±s,n = 3)Table 5 Effect of dispersed phase volume on MA-SLN comprehensive score ( ±s,n = 3)
分散相体积/mL 包封率/% 包封率评分 粒径/nm 粒径评分 ζ电位/mV ζ电位评分 综合评分 15 25.03±1.50 2.00 418.07±9.01 2.33 -16.27±1.29 3.00 2.27 25 37.23±1.79 3.00 327.57±8.92 4.67 -28.53±2.06 5.33 3.80 35 51.26±2.46 4.67 248.60±7.13 6.67 -37.83±1.47 7.00 5.53 45 57.33±4.67 5.33 197.20±4.35 8.00 -37.23±1.46 6.67 6.13 55 51.67±0.97 5.00 274.53±7.69 6.00 -34.90±1.48 6.33 5.47
由表4、5可知,当搅拌速度低于800 r/min时,由于无法为乳化体系的形成提供足够的能量因而MA-SLN成型与性能不佳,而搅拌速度过高(大于1000 r/min)会由于速度过大而增加微粒的动能、增大碰撞合并机率而影响微粒制剂的成型。乳化体系的形成是油相由整体逐渐变为微粒的渐进过程,因此需要一定的时间,当搅拌时间低于30 min时导致乳化不足而粒径过大,且微粒的形状不规整;而搅拌时间过长,高于30 min则由于增大微粒碰撞、合并机率而影响成型与粒径。研究表明,稀释相体积对MA-SLN的粒径影响较大,其原因是当稀释相体积过小时,单位体积内微粒的浓度增大,因此增大了微粒的碰撞机率增加聚集程度而使粒径变大;稀释相体积过大时,如大于45 mL时,则由于微粒稀释程度较大,得到的分散液固体含量过低,增加药物泄露且后续处理困难。因此,综合分析得到最佳制备工艺为搅拌速度为1000 r/min、搅拌时间为30 min与稀释相相体积为45 mL。
2.5 MA-SLN制备工艺和处方的确定
按照上述处方和工艺考察结果制备MA-SLN。称取苦参碱50 mg,单硬脂酸甘油脂0.15 g和大豆卵磷脂0.3 g,并加入5 mL无水乙醇作为油相;另取0.2 g普朗尼克F-68,加入纯化水10 mL作为水相;油相与水相分别置于65 ℃水浴中,使溶解;并于1000 r/min条件下将水相缓慢加入油相,搅拌乳化30 min,高速匀质4 min后将乳化体系分散到45 mL冷水中,300 r/min条件下搅拌固化20 min,过0.45 µm微孔滤膜,即得,于4 ℃存放,备用。
制备6批MA-SLN,其外观均匀一致无差异,MA-SLN粒径密度分布图和电势分布图见图2。测得其包封率为(56.12±0.82)%;平均粒径为 (196.31±6.26)nm,多分散指数(polydispersity index,PDI)为0.184±0.020;ζ电位为(-37.18±2.36)mV。表明本实验优化的制剂处方与制备工艺稳定、重现性高。研究表明[18],当微粒表面电位的绝对值大于30 mV时,微粒间由于存在较大的静电斥力而有利于制剂稳定性的提高并使粒径保持均匀,因此,MA-SLN具有较高的稳定性。
2.6 MA-SLN药剂学性质考察
2.6.1 MA-SLN形态观察 将MA-SLN样品适量,按照SEM和TEM的常规操作处理后,观察其外观形态结构。结果如图3所示,MA-SLN为规整平滑的均匀实体骨架球状结构,球体表面无苦参碱的规则晶体析出、内部显示均匀一致,表明苦参碱与SLN的骨架材料相容性良好而使苦参碱在MA-SLN呈现均匀分布,这种分布行为利于提高药物的稳定性和载药量。
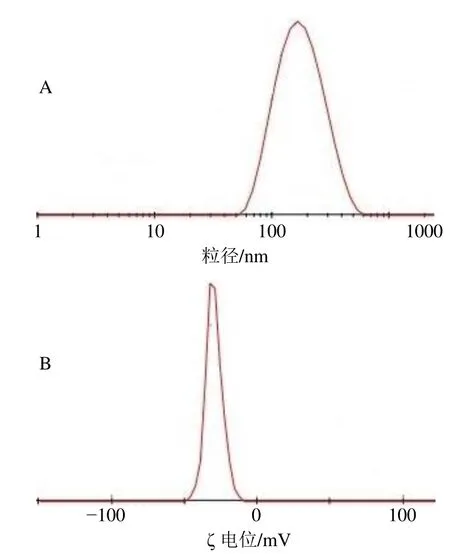
图2 MA-SLN粒径密度分布 (A) 和电势分布 (B)Fig.2 Particle size distribution by intensity (A) and ζ potential distribution (B)

图3 MA-SLN扫描电镜图 (×2400,A) 和透射电镜图 (×6000,B)Fig.3 SEM image (× 2400,A) and TEM images (× 6000,B) of MA-SLN
2.6.2 苦参碱包裹情况考察
(1)DSC扫描:分别取适量MA-SLN、B-SLN、苦参碱粉末、苦参碱与SLN物理混合物用DSC扫描检测苦参碱及载体特征峰的变化情况。DSC扫描条件为99.99% N2,升温速度20 ℃/min,温度范围0~280 ℃,结果如图4。由图4可知,苦参碱在70~78 ℃出现特征峰,B-SLN在55 ℃左右出现特征峰,物理混合物苦参碱+SLN分别出现了SLN和苦参碱的特征峰;而将苦参碱制成MA-SLN后,苦参碱的特征峰消失,仅显示SLN的特征峰。

图4 MA-SLN的DSC扫描分析图Fig.4 Analysis diagram of MA-SLN by DSC scan
(2)XRD分析:用XRD测定苦参碱粉末、MA-SLN和B-SLN。样品在45 kV、40 mA的Cu-Kα辐射下进行分析,测试范围5°~90°,扫描速率为 2°/min。扫描结果见图5。由图5可知,纯药物苦参碱的XRD谱图在6°~25°扫描角度之间显示出许多尖峰,表明苦参碱具有高结晶性,而MA-SLN则没有了药物苦参碱的主峰,结晶度降低;与B-SLN相比,MA-SLN的主峰没有移动,整体强度变化不大,在7°扫描角度处强度略有提高,这可能是由于脂质纳米粒中苦参碱的加入,导致了包载苦参碱的SLN结晶度发生变化。结合DSC扫描结果推断,苦参碱可被充分包裹到SLN中的实体骨架中,且苦参碱与SLN骨架材料相容性良好。
2.7 MA-SLN体外释药特点
图5 MA-SLN的XRD扫描分析图Fig.5 Analysis diagram of MA-SLN by XRD
采用透析法考察MA-SLN的释药特点,精密称取MA-SLN 2 mL,放入截留相对分子质量为2500的透析袋中,扎紧,置于50 mL的生理盐水溶液中,调整介质37 ℃、转速150 r/min进行体外透析试验,取样时间为1、2、3、4、6、9、12、18、24 h,取样量为3 mL,并补充同温等量的生理盐水,同法以相同质量浓度的苦参碱水溶液作为对照,计算累积释放率。结果见图6。由图6可知,苦参碱水溶液中苦参碱可快速释药,并在6 h后达基本释放完药物到平衡。而MA-SLN较苦参碱水溶液体外透析释药过程缓慢,在24 h后才逐渐达到平衡,因此MA-SLN体外释药具有缓释效果。

图6 苦参碱水溶液和MA-SLN体外释放曲线 ( ±s,n = 3)Fig.6 In vitro release of WMA and MA-SLN ( ±s,n = 3)
2.8 MA-SLN经皮渗透研究
取昆明种小鼠,脱颈处死后剥离皮肤,剪净皮肤上的毛,小心剔除皮下黏膜膜和脂肪组织,用生理盐水冲洗干净后切成小块(3.0 cm×3.0 cm),-20 ℃冷冻保存,1周内使用。
将角质层向上固定于立式双室扩散池上,在接收池加入7 mL生理盐水作为接收液,保持恒温在(32.0±0.5)℃,搅拌速度300 r/min;在供给池中分别加入相同苦参碱量的MA-SLN和苦参碱水溶液。分别在1、2、3、4、6、9、12、18、24 h取接收液0.5 mL,并补加0.5 mL新鲜接收液,将取得样品经0.22 μm微孔滤膜。按照“2.2.2”项下色谱条件测定苦参碱含量,按公式计算各时间点苦参碱累积透过量(ΔM)[19]。

C为接收液中的药物质量浓度(µg/mL),V为接收液的体积(mL),Ae为扩散池的有效面积(cm2)

图7 苦参碱累积透过量 ( ±s,n = 3)Fig.7 Cumulative permeation of matrine ( ±s,n = 3)
结果如图7所示。由图可知MA-SLN较苦参碱经皮渗透释药快,1~9 h时ΔM达到500 μg/cm2左右,是苦参碱水溶液的3.93倍(t检验,P<0.05),表明MA-SLN能够显著促进苦参碱的经皮渗透,其原因可能是MA-SLN以其较小的粒径(197 nm)和 亲脂性而增强了苦参碱的经皮渗透性,同时,药物在MA-SLN微粒中以高浓度的分子状态分布增大了其热力学活度,进一步利于苦参碱的经皮渗透吸收。9~24 h,MA-SLN可以持续稳定释放药物,以维持药物的经皮渗透吸收,表明MA-SLN具有良好的缓释行为;而水溶液中药物虽然也随时间延长而ΔM缓慢增加,但是增加不明显(t检查,P>0.05),表明苦参碱在苦参碱水溶液中难于经皮渗透吸收。结果表明MA-SLN不仅可以提高药物经皮渗透吸收的效率,还具有良好的缓释行为。
3 讨论
苦参碱具有多种药理作用,是一种具有巨大临床应用价值的天然药物。但是,由于苦参碱存在强烈持久的苦味且对胃肠道黏膜存在刺激,加之其生物半衰期较短,血浆清除快,临床为保证疗效需大剂量给药,进而增加不良反应发生频次,这些限制了苦参碱口服给药与注射给药的广泛应用。
本实验研究了以微乳-低温固化法制备MA- SLN,并对其药剂学性质与透皮给药行为进行了考察。研究发现,采用微乳-低温固化法制备MA-SLN,其工艺简单、周期短、成本低与可控性高,因此易于产业化放大。
实验中,为了准确测定苦参碱固体脂质纳米粒的包封率,必须将纳米粒和游离的药物有效分离。常用葡聚糖凝胶柱色谱法、透析法和超速离心法等分离方法。其中透析法过程缓慢,药物容易泄露;实验过程中也采用过超速离心法,但实验不易产生沉淀,且结果不稳定;而鱼精蛋白凝聚法主要是通过在纳米粒表面吸附某种蛋白质来增加固体脂质纳米粒的密度使游离药物与纳米粒通过离心后达到有效分离。本实验用约含2/3以上精氨酸的鲑鱼精蛋白,一种含有胍基的带正电的碱性氨基酸。苦参碱固体脂质纳米粒带有负电荷。多聚阳离子鱼精蛋白会与带负电的纳米粒产生絮凝作用,并通过离心将纳米粒和游离药物分离,该法简单易行,且分离效果较好。
本实验制备的MA-SLN呈规整平滑的均匀实体骨架球状结构,苦参碱在SLN均匀分布且二者相容性良好,MA-SLN对苦参碱具有较高的包封率(56.12±0.82)%,因此载药量大,可满足不同疾病治疗的需求;MA-SLN的平均粒径为197 nm(PDI为0.184),电位为(-32.0±2.1)mV,表明MA-SLN粒径分布均匀并具有良好的稳定性。MA-SLN具有较小的粒径,并且由于SLN的亲脂性材料将药物包裹于脂质结构中,增加了药物的稳定性和细胞透过率,因此,可以极大地促进苦参碱的透皮吸收[20-21],9 h时ΔM达到500 μg/cm2是苦参碱水溶液的3.93倍(t检验,P<0.05),同时,由于苦参碱被充分包埋于MA-SLN的实体骨架材料中因而具有较好的缓释行为,可持续释药24 h。MA-SLN为苦参碱提供了一种基于纳米微粒的新型外用缓释给药系统,在一定程度上促进了中药的传承与创新,因此,MA-SLN具有广阔的应用前景。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
食品安全导刊(2021年20期)2021-11-28
中华环境(2021年9期)2021-10-14
中华环境(2021年8期)2021-10-13
中华环境(2021年7期)2021-08-14
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年5期)2018-06-13
疯狂英语·新悦读(2017年6期)2017-06-24
电镀与环保(2016年2期)2017-01-20
现代工业经济和信息化(2016年12期)2016-05-17
中国民族医药杂志(2016年5期)2016-05-09
