
表面等离激元光催化固氮研究进展
2021-09-18 08:20江瑞斌王宇阳马丽霞
中国材料进展 2021年7期
江瑞斌,王宇阳,马丽霞
(陕西师范大学材料科学与工程学院 陕西省能源新材料与器件重点实验室,陕西 西安 710119)
1 前 言
氮是生物体合成氨基酸和核酸必需的元素,大气中虽然含有78%(体积分数)的氮气,但是由于其强的化学三键,无法直接被生物体利用,必须先转化为氨或氮氧化物才能被生物体利用。在自然界中,有两种途径实现固氮,一种是闪电固氮,另一种是微生物固氮。在闪电固氮中,闪电可以使氮气电离生成自由基,氮自由基随后可以和空气中的水成键,生成硝酸或者氨,而后随着雨水进入土壤中被生物体利用[1, 2]。微生物固氮中,一些细菌可以分泌固氮酶,在固氮酶的催化作用下将氮气转化为氨[3]。自然界中这两种固氮方式是早期生物体氮的主要来源,但是随着人口增加,这两种方式所提供的氮源不足以解决人类的温饱,因此需要发展人工固氮技术。
目前,工业上最重要的人工固氮技术是Haber-Bosch氨合成方法,该方法在高温(400~500 ℃)、高压(15~25 MPa)条件下将氮气和氢气转化为氨气[4, 5]。Haber-Bosch方法虽然实现了高效的氨合成,但是需要在高温高压条件下进行,且需要高纯的氢气,该方法的耗能占全球能耗的1%~2%,CO2排放量大约占全球总排放量的1%[6-8]。随着人类社会的发展,能源危机和环境污染日益严重,迫使人们发展新型的人工固氮技术。
近年来,人们在实验室发展了多种绿色固氮技术,包括电催化固氮[9, 10]、光催化固氮[11-14]、光电催化固氮[5, 15]。其中,光催化固氮被认为是一种极具前景的人工固氮技术,这是因为太阳辐射到地球上的能量高达120 PW,比人类能量总需求大几个数量级,完全可以满足人类需求[16]。在光催化固氮过程中,利用太阳光在温和条件下将氮气通过N2+3H2O=2NH3+3/2O2反应还原为氨,或通过2N2+5O2+2H2O=4HNO3反应氧化为硝酸。光催化固氮最早可追溯到,1977年Schrauzer和Guth利用TiO2的光催化作用实现了氮气转化为氨和N2H4,并且发现Fe2O3的负载可以提高TiO2光催化固氮活性[17]。从此,氧化物[18, 19]、硫化物[20, 21]、卤氧铋[22, 23]等各种半导体材料被用于光催化固氮,并使光催化固氮的效率得到了较大的提升。然而,半导体的光响应受其带隙的限制,很难实现对光的宽谱响应。和半导体不同,局域表面等离激元(localized surface plasmon, LSP)共振波长可以通过纳米颗粒尺寸和长径比来调控[24],能够实现对太阳光的宽谱响应和强吸收,因此已被应用于光催化分解水、光催化CO2还原和光催化固氮等反应中[25-27]。
本文将对近年来LSP光催化固氮的研究成果进行综述。首先对光催化固氮的基本原理进行介绍;随后对LSP的性质和其他光催化机制进行深入阐述;紧接着对近年来LSP光催化固氮的研究进展进行概括性介绍;最后对LSP光催化固氮中存在的问题及未来的发展趋势进行分析和展望。
2 光催化固氮基本原理
光催化固氮过程和其他光催化过程类似,也包含4个基本步骤(图1),第一步为氮气在催化剂表面活性位点的吸附,第二步为催化剂吸收光产生光生电子空穴对,第三步为光生载流子分离迁移至催化活性中心位置,最后一步为光生电子和氢还原氮气生成氨,或者光生空穴和氧氧化氮气生成氮氧化物。与其他光催化反应不同,氮气的吸附步骤在光催化固氮中至关重要。这是因为氮气为非极性分子,很难在催化剂表面吸附;另外由于氮气的三键作用极强,只有吸附位点能够对氮气产生充分的活化,才能有效降低后续还原反应的能垒,提高还原反应速率。
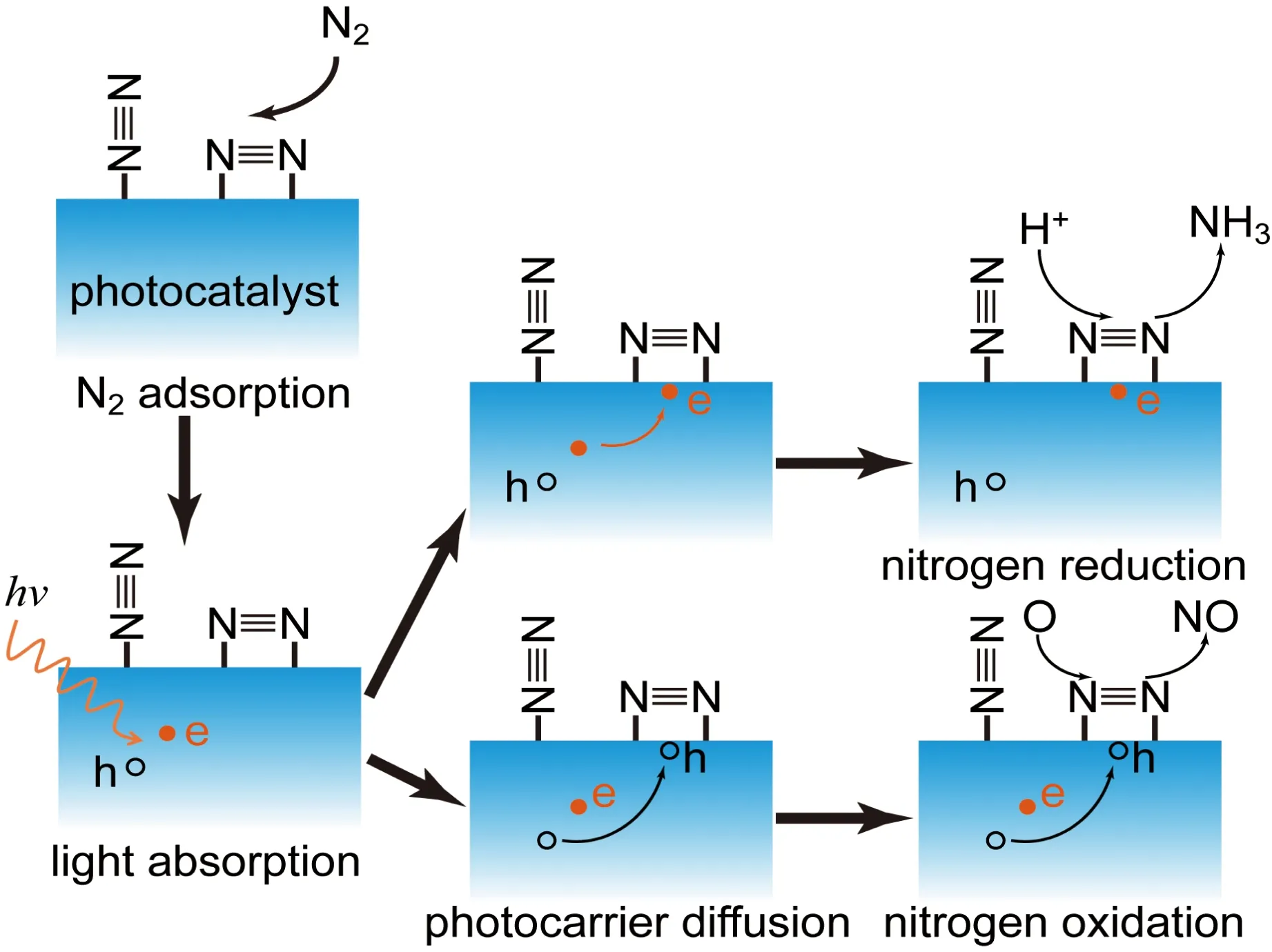
图1 光催化固氮过程Fig.1 Schematic of photocatalytic nitrogen fixation
2.1 氮气吸附和活化
由于氮原子的2s和2p轨道能量相近,当两个氮原子形成分子时,会发生sp轨道混杂,最终形成如图2a所示的分子轨道,最高占据轨道(HOMO)为弱成键2σ轨道,最低未占据轨道(LUMO)为反键轨道1π*轨道;2σ轨道和1π轨道共同形成了氮气分子中的三键[28]。由于强的共价三键和非极性特点,室温和常压下氮气的还原和氧化面临着以下挑战[2]:首先,氮气分子的键能高达941 kJ/mol,热力学上第一个键的断裂能高达410 kJ/mol,这使氮气三键的断裂十分困难;其次,氮气加一个氢原子是一个吸热过程,焓变为37.6 kJ/mol,表明氮气直接加氢在热力学上极为困难;另外,氮气具有负的电子亲合能(-1.9 eV)和极大的电离能(15.85 eV),使得氮气的活性极低;最后,氮气HOMO和LUMO轨道之间的能隙很大(~10.82 eV),使电子在两个能级上很难发生跃迁。因此,要使氮气的还原和氧化能够在室温和常压条件发生,就必须借助催化剂。

图2 氮气分子轨道(a)[28]; 过渡金属(transition metal, TM)原子氮气活化机制(b),主族元素(main group elements, MGE)对氮气活化机制(c)Fig.2 Molecular orbital of N2(a)[28]; Activation of N2 by TM (b), activation of N2 by MGE (c)
根据前线轨道理论,氮气吸附时主要通过2σ和1π*轨道和催化剂作用[29]。如图2b所示,当氮气和过渡金属(TM)活性位点相互作用时,会产生2σ轨道向TM的空d轨道给电子和TM的满d轨道向1π*轨道反给电子两种作用,这两种作用均会削弱氮气的三键,使氮气活化。当氮气和主族元素(MGE)活性位点相互作用时,氮气的2σ轨道会向主族元素的空p轨道或空sp杂化轨道给电子;同时,主族元素的满p轨道或满sp杂化轨道会向氮气的1π*给电子;这两种作用削弱氮气的三键,使氮气活化[30]。
2.2 光催化氮气还原
在光催化氮还原过程中,氮气捕获光生电子和氢反应生成氨气,与此同时,光生空穴氧化水或者牺牲剂。由于光催化氧化水产氧过程的动力学非常缓慢,通常为了加速空穴的消耗,提高光催化氮还原速率,需要加入空穴牺牲剂,例如甲醇、乙醇和Na2SO3等[11]。在没有牺牲剂的情况下,理想的光催化氮气还原是个六电子过程,涉及到的两个半反应及反应电位为[11]:
N2+6H++6e-→2NH3E0=0.55 V
(1)
3H2O+6h+→6H++3/2O2E0=1.23 V
(2)
N2+3H2O→2NH3+3/2O2ΔG0=7.03 eV
(3)
在热力学上,光催化反应的发生需要半导体的导带(CB)带边比还原反应的电位更负,而价带(VB)带边比氧化反应的电位更正。氮气还原生成NH3的平衡电位相对于标准氢电极为0.55 V(除了特殊说明,后边提到的电位均为相对于标准氢电极),生成NH4+的平衡电位为0.27 V,这两个反应的电位均比析氢反应的电位正,因此在热力学上更有利于发生氮还原反应(图3a);然而析氢反应是两电子过程,氮还原反应是一个六电子过程,因而在动力学上更有利于发生析氢反应。更为重要的是,氮气还原过程中的第一步加氢反应(N2+H++e-→N2H)极为困难,所需还原电位达-3.2 V,大多数半导体很难达到这一电位。幸运的是,当氮气被活性位点吸附活化后,这一步基元反应的还原电位明显升高。因此催化活性位点的设计在光催化固氮中就变得尤为重要,也成了光催化固氮研究的一个重点。另外,光催化氮还原还有可能通过四电子反应生成N2H4,该反应的电位为-0.36 V,低于生成氨的电位,因而在热力学上氮还原更有利于氨生成。
如图3b所示,氮气在催化剂上还原生成氨可以通过3种典型的反应路径。第一种为解离还原机制,在该机制中,N≡≡N三键在催化剂上先断裂生成两个N原子,随后N原子加氢还原成氨气。由于N≡≡N三键断裂非常困难,这种机制只在很少的光催化氮还原中观察到[31]。第二种机制为交替加氢机制,在该机制中,在两个N原子上交替加氢到N2H4,随后加氢的N—N键断裂生成氨气;若氮气是通过两个N原子同时吸附在催化上时,该机制又被称作酶催化机制。第三种机制为末端机制,在该机制中,先在一个N原子上连续加氢还原生成氨气,随后在另一个N原子上加氢生成氨气。由于后两种反应路径中,在氨气生成之前,N—N键一直没有断裂,因此后两种反应路径又被称为非解离反应路径。
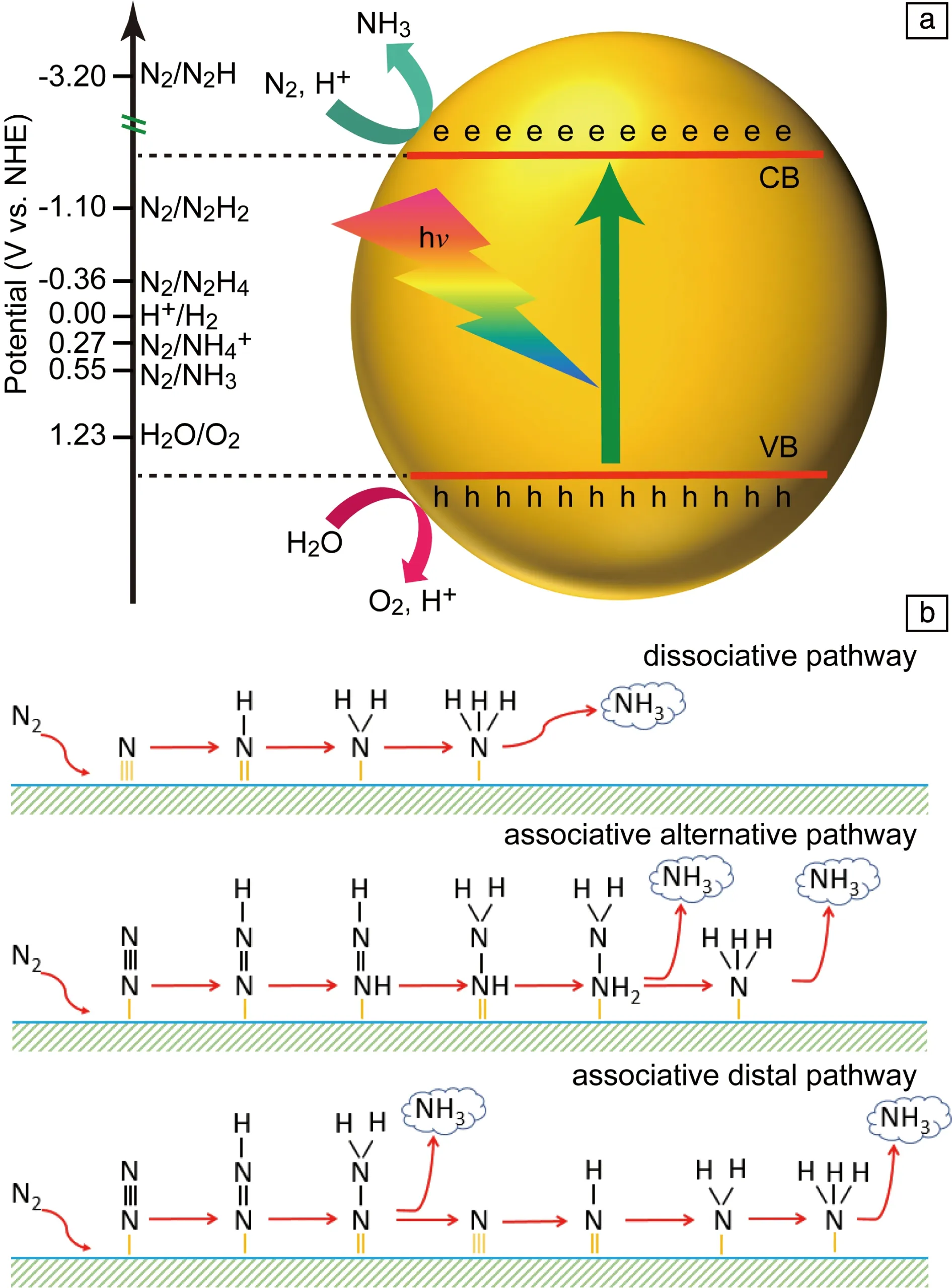
图3 氮气还原反应的氧化还原电位(a)[11];3种典型氮气还原生成氨的路径(b)Fig.3 Equilibrium potential of nitrogen reduction (a)[11]; Three typical nitrogen reduction paths (b)
2.3 光催化氮气氧化
在光催化氧化固氮过程中,氮气失去电子被氧化成NO,进一步被氧化生成NO3-[19, 32, 33]。根据氧化产物中氧的来源,可以将光催化氮气氧化分成CB和VB两种机制。如图4所示,在CB机制中,O2吸附在催化剂表面并捕获光生电子,形成超氧自由基,进而O—O键断裂形成两个吸附的O原子,随后N2分子吸附在这两个O原子上,最后N—N键断裂生成NO。在VB机制中,两个吸附在催化剂表面的水分子捕获光生空穴,被氧化生成两个吸附的·OH和H+;两个吸附的·OH进一步被光生空穴氧化生成两个吸附的O原子,随后两个O原子和N2反应生成NO。两个机制中所生成的NO进一步与水和O2反应生成HNO3,整个过程包含以下反应:
N2+O2→2NO
(4)
N2+2H2O+4h+→2NO+4H+
(5)
4NO+2H2O+3O2→4HNO3
(6)
与光催化氮气还原相比,关于光催化氮气氧化的报道较少,但是根据上面提到的光催化氮气氧化机理,可以预测,通过对光催化剂进行修饰改性,可以提高光催化氮气氧化效率。
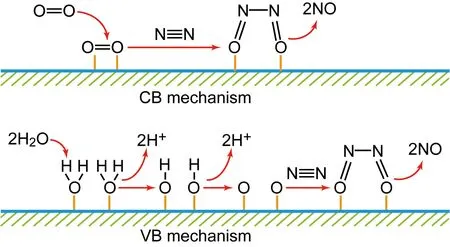
图4 两种氮气氧化生成NO的机制Fig.4 Two mechanisms of nitrogen being oxidized to NO
3 局域表面等离激元
表面等离激元是金属(或高掺杂半导体)中自由载流子在外电磁场的激发下,载流子运动和电磁场相互激励产生的共谐振荡。类似于声子是晶体中原子集体振荡运动的量子化描述,表面等离激元(或称为电浆子)是载流子及电磁场的共谐振荡量子化后的准粒子。表面等离激元携带相应的准动量和能量。表面等离激元主要有两类,一类为共谐振荡沿着金属薄膜和介质的界面传播,称为传导表面等离激元(surface propagating plasmon,SPP);另一类为共谐振荡限制在纳米颗粒的边界上,称为局域表面等离激元(localized surface plasmon,LSP)。
3.1 局域表面等离激元特性
LSP可以采用量子描述,也可以采用经典电动力学描述。在经典物理中,纳米结构中自由电子气在外加电场的作用下会相对于离子实发生移动,移动的方向随着电磁波中电场方向的改变而改变(图5a)。在一定环境中,一个纳米结构可以有多个固有的电子振荡频率。当外加电磁波的频率和固有电子振荡频率相同时会发生共振,这时电子振动的振幅达到最大值,即产生了LSP共振。在共振激发时,纳米结构的吸收和散射截面积、结构周围的局域电磁场都被极大地增强。根据经典电动力学,可知LSP共振波长、光学截面积和电磁场增强等性质依赖于纳米结构的材料、尺寸、形状和结晶性以及周围环境[34, 35]。对材料的依赖性是因为自由载流子的密度和载流子的迁移率均由材料决定。在可见和近红外波段产生LSP共振,材料的自由载流子的密度必须达到1021cm-3[36],因此Au,Ag和Cu是能够在可见和近红外波段产生LSP共振的优异材料。纳米结构的尺寸和形状影响自由载流子和离子实之间的恢复力以及自由载流子在纳米结构表面的分布,因而会影响LSP共振性质。晶界会对运动的自由载流子产生散射作用,从而破坏共振的相干性,因此会影响LSP共振性质。
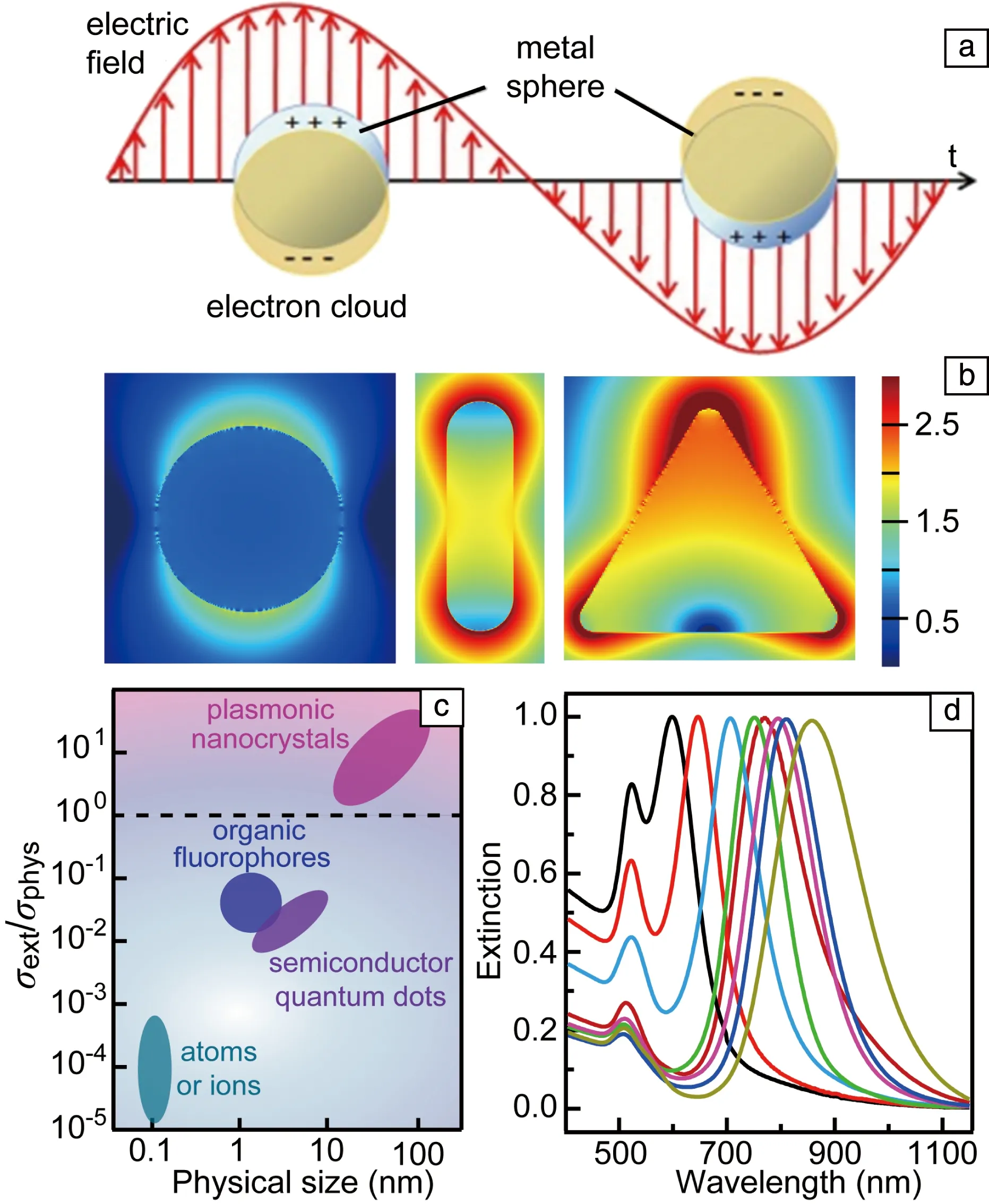
图5 局域表面等离激元(localized surface plasmon,LSP)共振示意图(a),场增强效应(b)[37],不同纳米光学物种的尺寸及消光和物理截面积比值(c)[38],不同长径比金纳米棒的消光光谱(d)[39]Fig.5 LSP resonance (a), field enhancement effect (b)[37], ratio of optical and physical cross section of different optical species (c)[38], extinction spectra of different gold nanorods (d)[39]
周围环境对LSP共振性质的影响可以从周围介质的极化角度进行理解,LSP共振会引起周围介质的极化,极化的介质反过来会对载流子振荡产生额外的作用力,影响共振;不同介质极化能力不同,产生的额外作用力则不同,从而周围介质影响LSP共振性质。
在共振激发时,LSP使纳米结构表现出一系列新奇的特性。首先LSP可以打破衍射极限,将光局域到纳米尺寸,使纳米结构表面附近的电场强度比入射光中的电场强几个数量级(图5b)[37]。LSP的另一个重要的性质是可以使纳米颗粒具有极大的光学截面积,在所有的发光体中只有LSP纳米颗粒的光学截面积大于其物理截面积(图5c)[38],因此LSP纳米颗粒和光具有极强的相互作用。另外,对于同一材料构成的纳米结构,LSP共振波长可以通过调控纳米结构的形状和尺寸在很宽的波长范围内进行调节,因此能够实现对太阳光的宽谱响应。例如,图5d为金纳米棒的LSP消光谱,通过调控纳米棒的长径比,LSP共振波长可以从500 nm调控到近红外甚至中红外波段[24, 39]。
如图6a所示,LSP在激发后通过辐射和非辐射两种方式快速衰减,LSP的寿命只有几飞秒[40]。在辐射衰减中,LSP发射出光子。在非辐射衰减中,LSP通过带内和带间跃迁生成电子-空穴对[40]。由于生成的电子和空穴的能量高于平衡态电子和空穴的能量,因此被称为热载流子(hot carriers);同时,由于其能量较高,热载流子在光催化和光伏等领域具有巨大的应用前景。在随后的100 fs~1 ps内[41],热电子通过电子间的散射作用使电子之间达到热平衡,这时整个电子呈现费米-狄拉克分布(图6b),但是整个自由电子的温度高于晶格温度。这时的电子有时也被称为热电子,但是电子能量远低于LSP衰减直接生成的热电子的能量,为了区分这两种电子,达到热平衡的电子应更严谨地被称为温电子(thermal electrons)。在随后的几百飞秒到几皮秒,通过电子和声子相互作用将能量转化为晶格振动能,实现光热转化。由于极大的吸收截面积,LSP纳米颗粒具有优异的光热效应,该效应常被用于光热催化和肿瘤的光热治疗中。
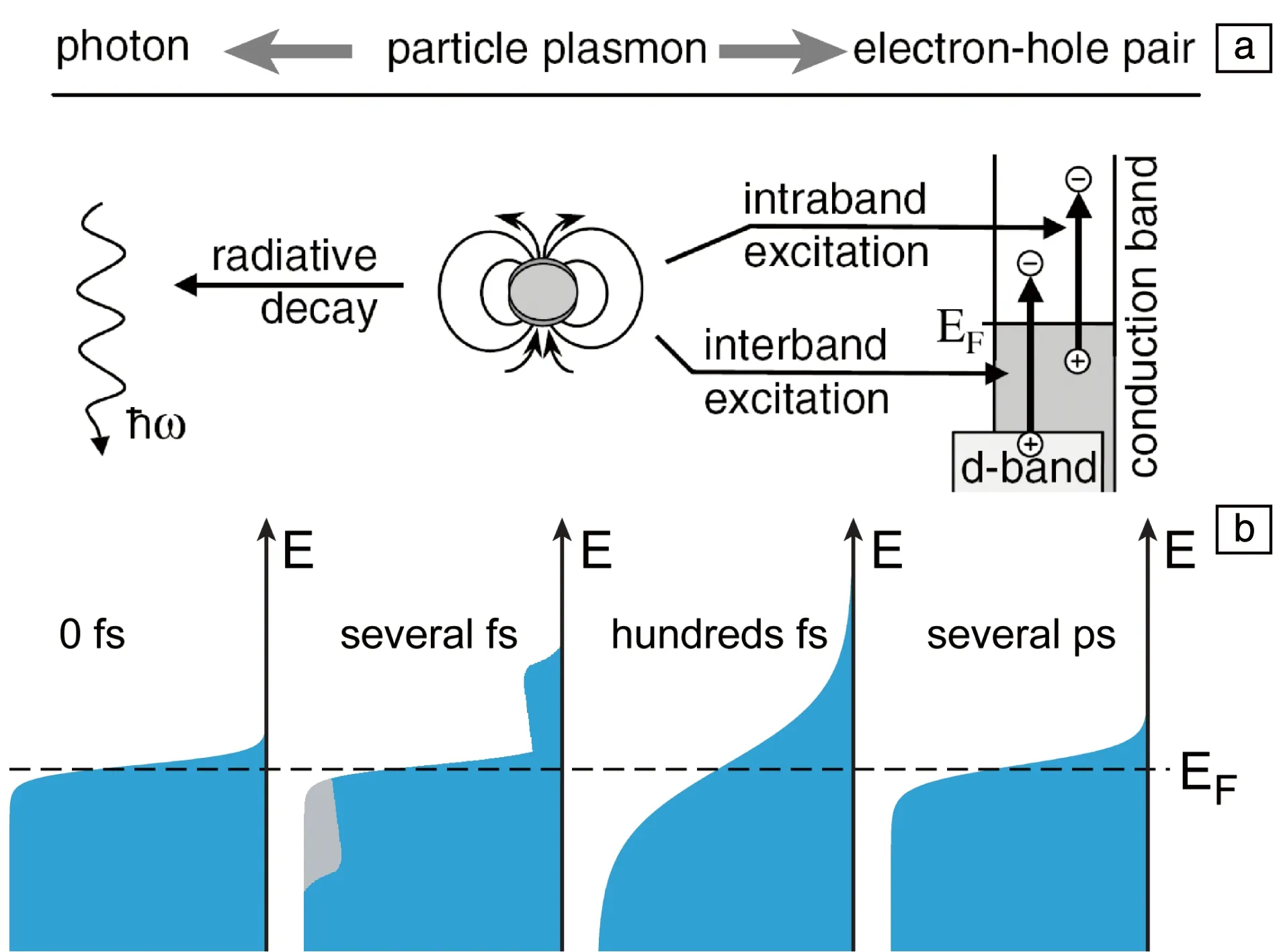
图6 LSP衰减过程[40]:(a)LSP衰减方式,(b)电子分布随时间的变化Fig.6 Decay of LSP[40]: (a) decay channels of LSP, (b) distribution of electrons at different time during LSP decay
3.2 局域表面等离激元的光催化机制
目前提出的LSP增强光催化的机制主要有场增强效应、热电子效应和光热效应3种,其中场增强效应直接来自LSP共振,而后两种是LSP非辐射衰减产生的效应。前面提到,当LSP共振激发时,可以在纳米颗粒表面产生极强的电场增强效应,而增强的电场可以增加和LSP具有相同波长的光学跃迁过程[25]。因此,当LSP纳米颗粒和半导体结合在一起时,LSP共振的场增强效应就会增加半导体对光的吸收,从而提高半导体的光催化效率(图7a)。需要说明的是,利用场增强效应增加半导体的光吸收,需要LSP的共振能量高于半导体的带隙,因此场增强效应无法拓宽半导体的光响应波长范围。另外,还有一种利用场增强效应增强光催化的机制是,利用增强的电场与分子中极性化学键的偶极矩相互作用,增加极性化学键的振动,使分子中化学键活化(图7b)[42];由于该增强机制是通过电场和极性键的偶极矩作用来实现,因此不能增强非极性键的活化。
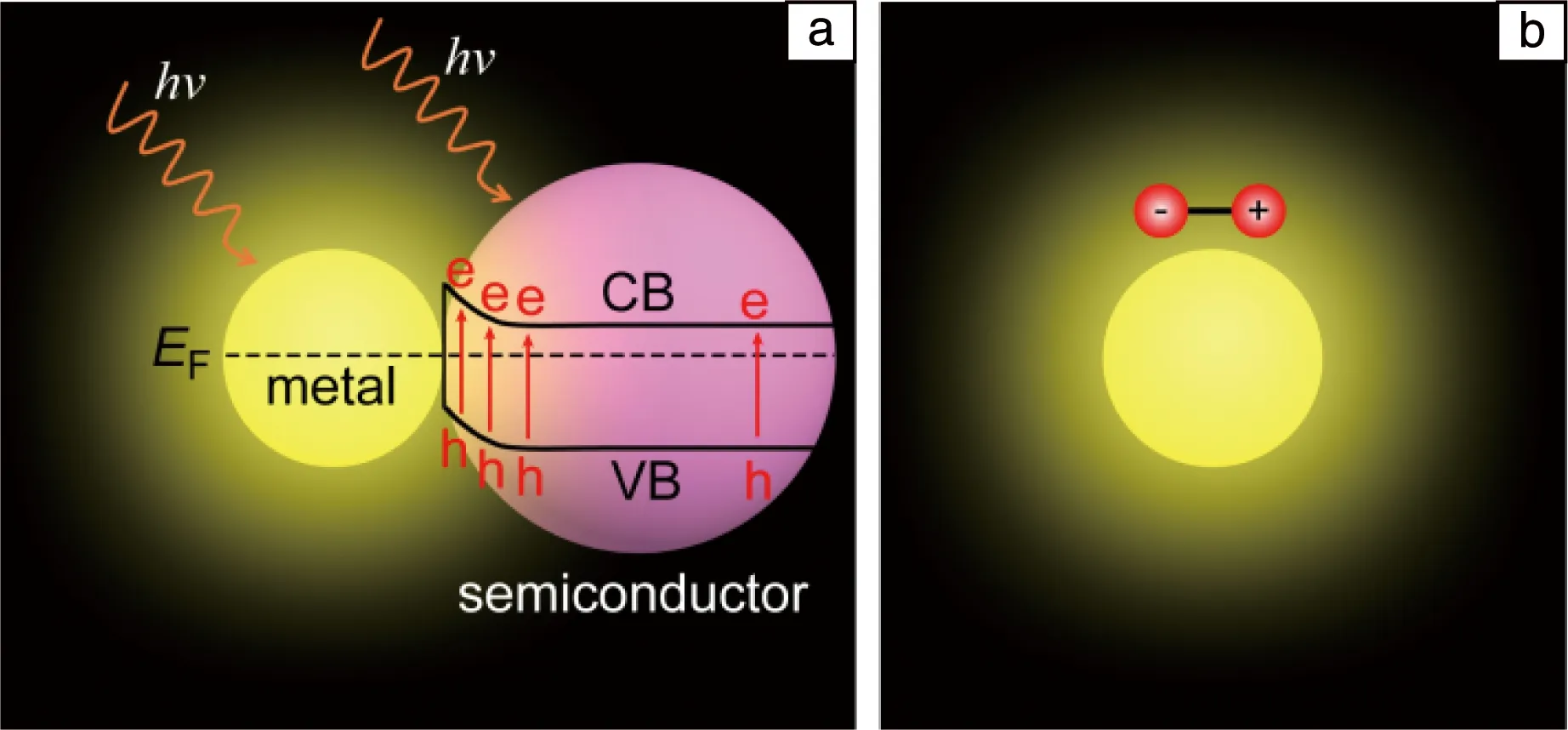
图7 LSP增强半导体光吸收(a)和增强分子极化(b)的示意图Fig.7 Schematics of LSP-enhanced light absorption of semiconductors (a) and LSP-enhanced the polarization of molecules (b)
前面提到LSP在衰减过程中会产生热载流子,LSP热载流子光催化机制正是通过LSP的高能热载流子转移到反应物,致使催化反应发生[25]。在该机制中,热载流子直接由LSP衰减产生,因此该机制使光催化不再受半导体带隙的限制,而是由LSP共振波长决定。通过调控纳米结构的尺寸和形状,可以使LSP共振波长覆盖很宽的光谱范围,因此LSP热载流子光催化机制可以实现宽谱光催化,使近红外光催化成为可能[25]。同时,LSP使纳米结构具有极大的光学截面积,可以实现对光的强吸收。因此,LSP热载流子机制使光催化可以实现宽谱响应和强吸收,从而提高光催化效率。
LSP热载流子光催化剂根据结构主要可以分为两类:一类是由LSP金属和半导体组成,另一类是纯金属结构。在LSP金属和半导体构成的结构中,热电子(空穴)先到达半导体的导带(价带),随后在半导体中参与催化反应。目前认为热载流子转移到半导体有两种机制,一种为间接转移机制,另一种为直接转移机制。在间接转移机制中,LSP在金属中先衰减生成热载流子,热载流子随后越过LSP金属和半导体之间的势垒,转移到半导体(图8a)。例如,对于n型半导体,热电子越过肖特基势垒进入半导体的导带。在该机制中,由于热电子需要越过界面处的势垒,只有那些能量高于界面势垒的热载流子才能到达半导体中;同时由于LSP金属中热电子的寿命小于100 fs,因此该机制中的热载流子效率较低。理论研究结果表明,在实际的材料中,该机制热电子效率的理论最大值为8%[43]。在直接转移机制中,LSP由于界面阻尼直接将电子从金属激发到半导体的导带中形成热电子(图8b)。该机制由界面阻尼引起,因此要求半导体和LSP金属有强的结合作用,在界面处LSP金属和半导体形成杂化的电子态。由于该机制有效避免了热电子在LSP金属中由于电子间相互作用导致的能量损耗,因而被认为具有更高的效率[44]。例如,研究发现,在Au/CdSe结构中,直接转移机制的热电子效率可达24%[45]。然而,目前的报道基本上都是采用间接转移机制解释LSP金属/半导体中的热电子光催化,这可能是因为目前报道的LSP金属/半导体热电子催化剂中大多数半导体为氧化物、卤氧化物和石墨相氮化碳(g-C3N4)等,这些半导体和LSP金属结合弱,很难形成界面电子态,因而不能实现热电子直接转移。要获得高效LSP热电子催化剂,未来研究就需要对LSP金属/半导体界面的电子结构进行精准设计和调控。
在LSP纯金属催化剂中,由于金属中价电子高度离域化的特点,无论是单纯的LSP金属,还是其它金属原子掺杂的LSP金属或者其它金属包覆的LSP金属中,LSP共振都是由其共同的价电子构成,且覆盖整个纳米结构。当分子吸附在LSP纯金属催化剂表面时,和LSP金属/半导体结构类似,热电子转移到分子的机制也存在间接和直接两种(图8c,8d),具体为哪种机制取决于分子和金属的轨道杂化程度,当杂化程度高,形成新的电子态时,则可以发生直接转移机制,否则为间接转移机制。同样,直接转移机制具有更高的热电子效率。

图8 热电子转移机制[44]:LSP金属/半导体结构中热电子间接转移机制(a)和直接转移机制(b),LSP金属/分子体系中热电子间接转移机制(c)和直接转移机制(d)Fig.8 Hot-electron transfer mechanism [44]: indirect (a) and direct (b) transfer mechanism in LSP/semiconductor structures, respectively; indirect (c) and direct (d) transfer mechanism in LSP/molecule systems, respectively
LSP衰减所产生的未发生转移的热电子,将通过快速的电子-电子和电子-声子相互作用将能量转移给晶格振动,产生光热效应[41]。由于化学反应的速率常数由阿伦尼乌斯公式决定,温度升高将加速化学反应。当LSP纳米颗粒分散在溶液中时,由于液体高的热扩散系数和对流作用,纳米颗粒表面的温度和溶液的温度相差很小(短脉冲强激光辐射除外),形成宏观上的热效应;而当LSP纳米颗粒分散在固体基底上时,由于气体的热扩散系数较小,纳米颗粒的温度将有可能远高于周围环境温度,形成局域热效应[46]。由于氮气还原生成氨气是一个放热反应,宏观加热不利于氨气生成,但是氮气又需要高温才能活化,因此LSP纳米颗粒的局域光热效应为提高氮气转化率提供了可能。利用局域高温活化氮气,而整个反应体系温度较低,可以有效增加氮气的转化率。
4 局域表面等离激元光催化固氮
尽管LSP金属/半导体结构在很早之前已经用于光催化固氮[17,47],但是这些研究没有考虑LSP效应,认为LSP金属只起到助催化剂的作用。近20年来,随着不断研究,人们对LSP有了深刻的认识,LSP近几年才被应用到光催化固氮中,LSP光催化固氮的研究还处在初级阶段。目前报道的LSP光催化固氮都是基于氮还原反应,还没有LSP光催化氮氧化的文献。根据前文的氮氧化的机理,理论上LSP光催化氮氧化也有可能发生。表1给出了近年来LSP光催化氮还原方面的研究成果,这些研究利用LSP均提高了光催化氮还原的效率。考虑到LSP催化剂的类型直接决定着LSP光催化氮还原的机制,下文将根据LSP催化剂的类型对LSP光催化固氮进行介绍;同时,考虑到在多数研究中,尽管纳米结构的材料组成不同,但是催化机制相同,下文将重点围绕LSP光催化氮还原机制展开,而非对LSP光催化固氮的研究成果逐篇列举。

表1 LSP光催化固氮研究成果
4.1 双金属表面等离激元固氮光催化剂
由于纯LSP金属(Au,Ag和Cu)的催化活性很弱,不能有效吸附和活化氮气,因此很难直接实现LSP光催化固氮,需要和其它氮气活化中心相结合才能够实现LSP光催化固氮。在双金属LSP固氮光催化剂中,LSP金属的主要作用是吸光和产生热电子,而另外一种金属的作用是吸附和活化氮气,因此另外一种金属必须和氮气具有强的相互作用。鉴于过渡金属Rh,Ru和Fe对氮气具有较高的催化活性,将其和LSP金属相结合可以实现LSP光催化固氮。Xiong等[31]采用Au/Ru核壳结构实现了LSP光催化氮还原。Au/Ru核壳结构采用一步高温湿化学还原法得到。研究发现,氨的生成速率随着Au/Ru结构中Ru含量的增加先增加而后减小(图9a),这是因为随着Ru的增加,氮气还原的活性位点增加,而LSP共振会减弱,导致光吸收减小。在Ru的含量(摩尔分数)为31%时,氨的生成速率达到最大值(101.4 μmol·g-1·h-1),是纯Au纳米球的7.3倍。Au/Ru固氮的表观量子效率(apparent quantum efficiency,AQE)随波长的变化和LSP吸收光谱一致(图9b),在550 nm处的AQE为0.17‰。对催化机理的研究表明,Ru是催化活性中心,且氮还原过程为解离还原机制,LSP提供还原电子。该研究虽然实现了双金属的LSP光催化氮还原,但是整个还原反应是在0.2 MPa纯氮气气氛下进行的,且光源较强(400 mW·cm-2),整体的AQE也较低。
随后,Wang等[53]采用CuFe合金结构也实现了LSP光催化氮还原。合金中Fe的含量通过酸刻蚀来控制,研究发现,随着Fe含量的减小,氨的生成速率先增加后减小(图9c)。Cu96Fe4具有最大的氨气生成速率342 μmol·g-1·h-1,而在Cu99.9Fe0.1上基本没有氨生成。Cu96Fe4固氮的AQE和其吸收光谱一致(图9d),其中在短波处有带间电子跃迁的贡献,在LSP共振波长535 nm处AQE为0.13%。进一步研究表明,Fe是氮气还原反应的活性位点,其中Fe二聚体具有更高的催化活性。整个反应机理为CuFe中的LSP产生热电子,Fe提供催化活性位点。
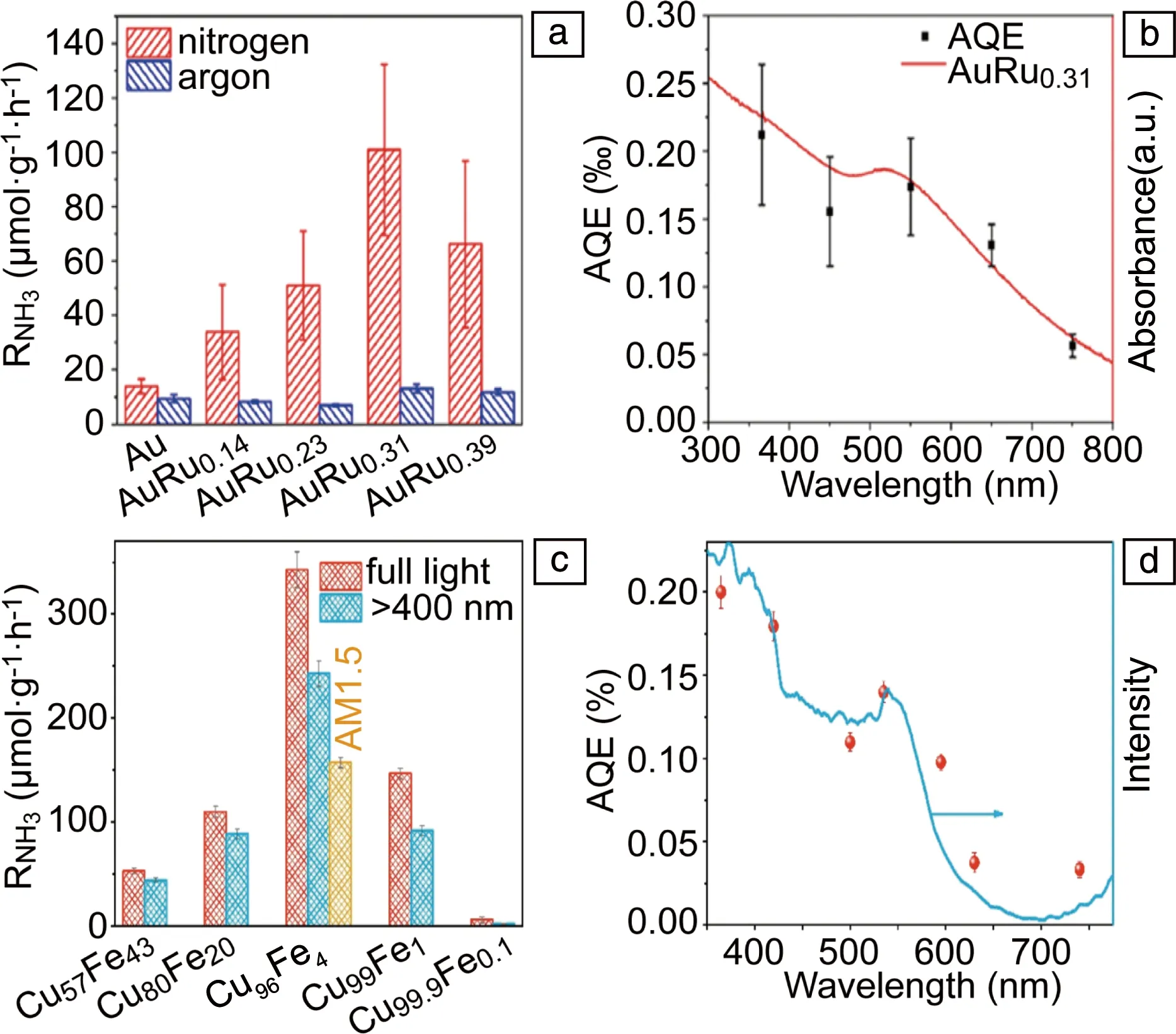
图9 不同Au/Ru样品的产氨速率(a),Au/Ru0.31的产氨表观量子效率(AQE)和吸收光谱(b)[31];不同CuFe样品的产氨速率(c),Cu96Fe4的产氨AQE和吸收光谱(d)[53]Fig.9 NH3 production rate on different Au/Ru samples (a), AQE of Au/Ru0.31 (b)[31]; NH3 production rate on different CuFe samples (c), AQE of Cu96Fe4 (d)[53]
以上是目前仅有的两篇采用纯金属结构实现了LSP光催化固氮的文献。与Au/Ru相比,CuFe的固氮效率明显地提高,这可能有几方面原因:首先Fe对氮气可能具有比Ru更高的催化活性;其次是CuFe中Fe可能以低聚体的形式存在,使其催化活性更高;另外是合金结构可能具有更高的热电子转移效率。由此可见,在未来研究中,通过在LSP颗粒表面精准设计和制备单原子或者低聚体氮气催化活性中心,有望获得更高的光催化固氮效率。
4.2 金属/半导体表面等离激元固氮光催化剂
金属/半导体结构是非常重要的一类LSP固氮光催化剂,该类LSP固氮催化剂可以利用LSP的场增强效应增强半导体的光吸收,提高光催化效率;同时也可以通过LSP热电子向半导体转移,实现光催化固氮;另外,LSP金属可以作为助催化剂促进半导体中载流子的分离,提高光催化效率,但是该作用与LSP无关,因此下文不再介绍。由于场增强效应不能拓宽半导体对光的波长响应范围,在光催化固氮中关注较少。目前仅有的一个利用场增强效应增强光催化固氮的研究工作是基于Au纳米球和碳点(CD)构成的结构[55],研究结果表明Au纳米球中LSP可以增加CD的光吸收,同时Au纳米球可以促进CD中载流子的分离,提高了光催化固氮效率。与场增强效应相比,金属/半导体中LSP热电子效应在光催化固氮中受到了较为广泛的关注。作者和合作者利用Au纳米球和含有氧空位(OV)的TiO2纳米片(TiO2-OV)之间的静电相互作用构建了Au/TiO2-OV结构,该结构的可见光固氮效率为79 μmol·g-1·h-1,在550 nm波长处的AQE为0.82%[56];进一步研究表明,氮气可以在TiO2纳米片中的OV上形成强的化学吸附(图10a),因此OV为氮气催化反应的活性位点;通过一系列对比实验发现,只有当Au和TiO2界面能够保证热电子顺利转移时,才能获得优异的固氮效率(图10b)。根据实验结果,提出了如图10c所示的“串级”LSP光催化固氮机制,在该机制中,LSP的作用是吸收光产生热电子,TiO2-OV的作用是提供催化活性位点,热电子由Au纳米球转移到TiO2的导带,然后被OV捕获将氮气还原。该机制成功将吸光和催化活性材料分开,增加了氮气催化活性材料设计的灵活性。在已报道的LSP固氮光催化剂中,许多都是基于此机制,例如Au/TiO2[50]、Au/Ag2O[52]、Au/(BiO)2CO3[54]、Au/g-C3N4[59, 60]、Ag/W18O49[61]、Au/CeO2[66]等。尽管这些工作采用LSP热电子机制在金属/半导体结构中实现光催化固氮,但是固氮效率依然较低,这主要有两方面原因:一方面,这些结构中的热电子为间接转移机制,导致热电子效率较低;另一方面,这些结构中的氮气活化位点主要为一些阴离子空位,对氮气的催化活性有限。在未来的研究中,可以通过界面结构调控改变热电子的转移机制,提高热电子效率;同时通过单原子的制备手段在半导体中引入对氮气具有更高催化活性的催化位点,有望获得更高的光催化固氮效率。
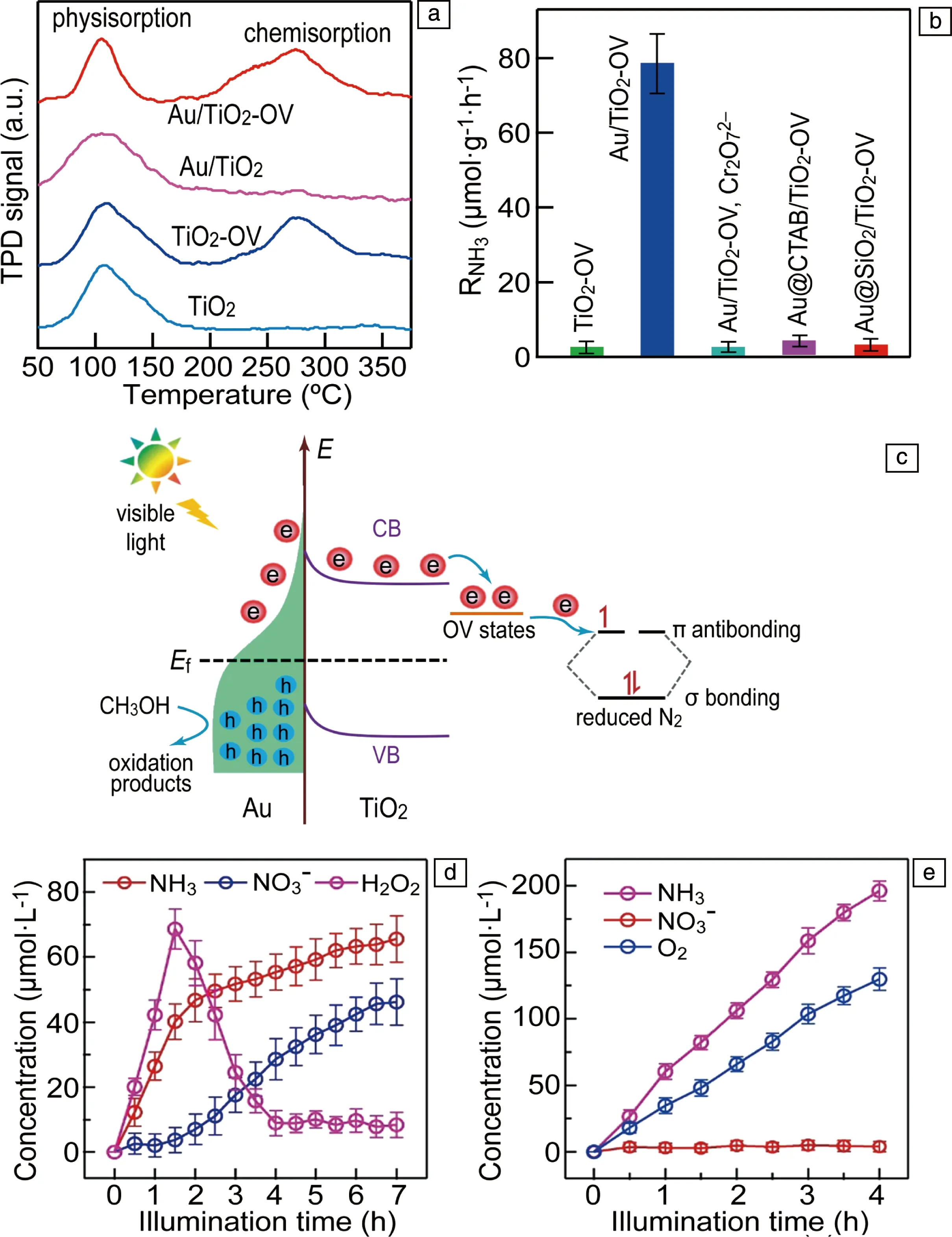
图10 不同样品上N2的程序控制升温脱附(temperature programmed desorption,TPD)信号(a),不同样品的产氨速率(b),Au/TiO2-OV光催化固氮机制(c)[56];Fe-TiO2光催化固氮过程中NH3、NO3-和H2O2浓度随时间的变化(d),Fe-TiO2/Au光催化固氮过程中NH3、NO3-和O2浓度随时间的变化(e)[67]Fig.10 N2 TPD profiles on different catalysts (a), NH3 production rate over different catalyst (b), nitrogen fixation mechanism on Au/TiO2-OV (c)[56]; Variation of NH3, NO3- and H2O2 concentrations during photocatalysis of Fe-TiO2 (d), variation of NH3, NO3- and O2 concentrations during photocatalysis of Au/Fe-TiO2 (e)[67]
除了增强光催化固氮效率外,LSP共振还可以改变光催化固氮的反应路径。作者和合作者研究发现,Fe掺杂的TiO2(Fe-TiO2)光催化固氮时,氮气会发生歧化反应生成NH3和NO3-(图10d);而当引入Au纳米颗粒时,光催化固氮产物只有NH3(图10e),表明LSP共振改变了固氮反应路径[67]。进一步研究发现,在Fe-TiO2光催化过程中有H2O2生成,当H2O2达到一定浓度后,就会将NH3氧化生成NO3-;当引入Au后,LSP共振产生的热空穴可以将H2O2快速氧化生成O2,避免了NH3被氧化。
另外,Zhang等[64, 65]利用LSP金属的光热效应和半导体的光催化性能实现了双温区光催化固氮,有效解决了氮气需要高温活化而低温有利于反应平衡向生成氨气方向移动的矛盾。在Fe/TiO2-xHy和Ru/TiO2-xHy结构中,利用Fe和Ru的LSP光热效应,在Fe和Ru颗粒处形成了局域高温区,使氮气活化,而随后的加氢还原反应在低温的TiO2-xHy上进行,从而有效提高了氨气的平衡浓度。尽管Fe和Ru不是理想的LSP材料,但Zhang等[64, 65]的研究结果为LSP固氮光催化剂的设计提供了新思路,在后续的研究中可以将对氮气具有高活化能力的金属(Fe,Ru和Rh等)生长在具有优异LSP共振性能的金属(Au,Ag,Cu和Al)颗粒表面,再将这种双金属结构与半导体相结合,有望进一步提高LSP光热固氮性能。
4.3 缺陷半导体表面等离激元固氮光催化剂
除了Au,Ag和Cu外,金属氮化物和高掺杂的半导体纳米结构中也可以产生LSP共振,同时由于半导体中的缺陷位可以作为氮气的催化活性中心,因此高掺杂的半导体纳米结构单独可以实现LSP光催化固氮。需要说明的是,高掺杂半导体的LSP光催化和传统的半导体光催化存在本质的区别。如图11a所示[63],传统的半导体光催化是半导体吸收能量大于带隙的光子,将电子从价带激发到导带,同时在价带形成空穴,电子和空穴随后参与还原和氧化反应。然而,当半导体的掺杂浓度很高时,半导体的导带(价带)中将会有大量的自由电子(空穴)填充,在光的激发下,这些载流子产生LSP共振,LSP衰减产生的热电子和空穴参与还原和氧化反应(图11b[63])。根据这一设想,Wang等[63]采用MoO3-x纳米片实现了可见光和近红外光催化固氮(图11c)。由于本征的MoO3的带隙大约为3.3 eV,无法实现可见和近红外光催化。而MoO3-x纳米片中存在大量的氧空位,使其在可见和近红外波段产生LSP共振,因而在可见和近红外波段具有光催化活性。同时,氧空位为氮气的吸附和活化提供了大量的活性中心。MoO3-x纳米片光催化固氮的AQE呈现出和LSP共振吸收谱相同的趋势,在800 nm波长处,AQE可达0.3%。该工作为新型LSP固氮光催化剂的设计提供一种新思路。
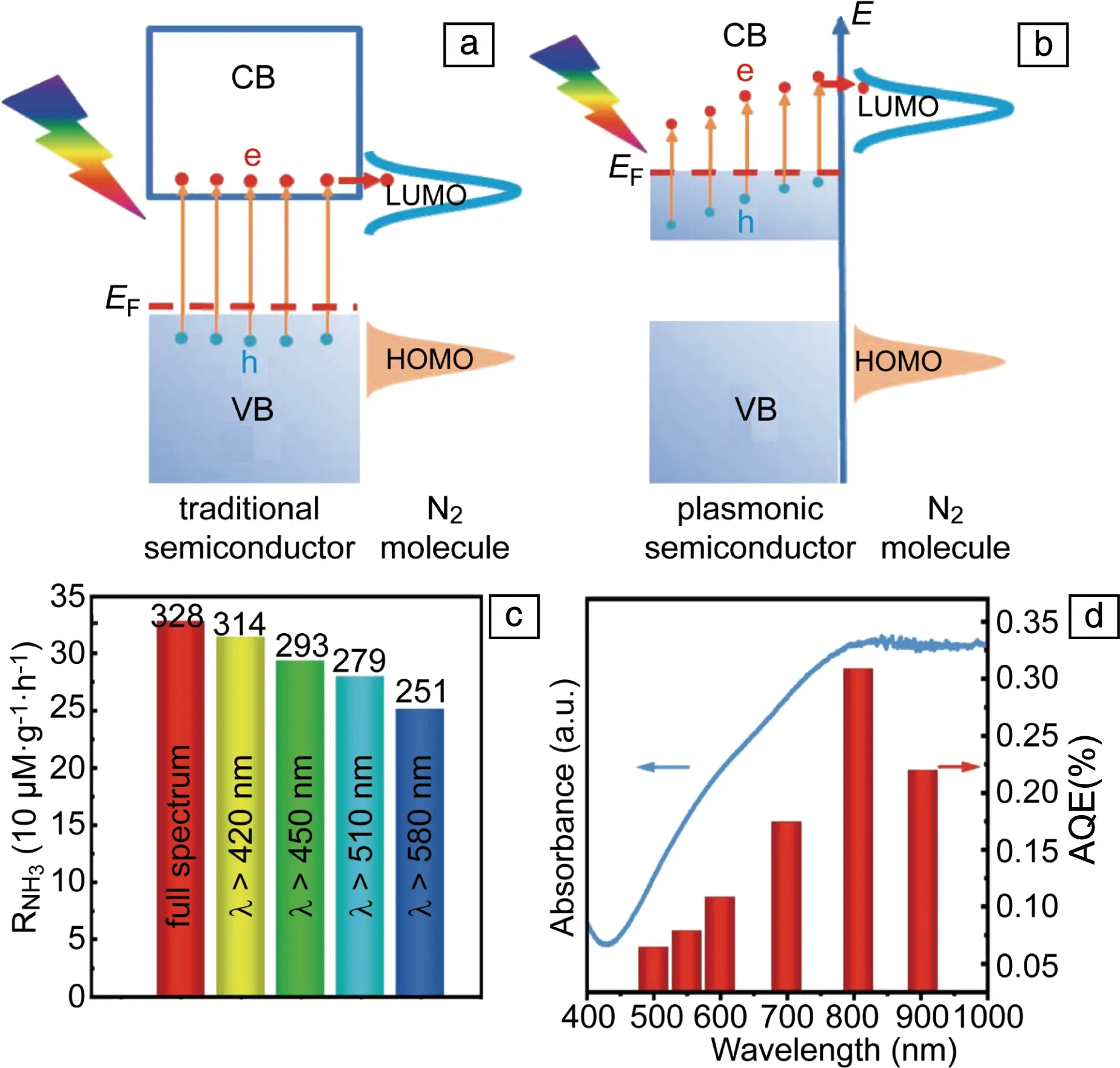
图11 传统半导体(a)和LSP半导体(b)光催化机理示意图[63]; MoO3-x在不同光谱下的产氨速率 (c),MoO3-x的产氨AQE和吸收光谱 (d)[63]Fig.11 Photocatalysis mechanisms schematics of traditional semiconductors (a) and LSP semiconductors (b) [63]; NH3 production rate on MoO3-x under different light illumination (c), AQE of MoO3-x (d)[63]
5 结 语
本文对局域表面等离激元(localized surface plasmon, LSP)光催化固氮的最近研究进展进行了综述,着重介绍了光催化固氮的基本原理、LSP的性质和其光催化机制以及近年来LSP光催化固氮方面取得的代表性进展。LSP光催化固氮作为一种新型的固氮技术,依然处在研究的初级阶段,要实现高效的LSP光催化固氮,在未来研究中需要在以下方面做出努力:
(1)建立标准的测试方法。目前LSP光催化固氮反应条件相差较大,例如光源功率和压力,导致催化结果不具有可对比性;另外,产氨速率的单位也不一致,例如有些文献为μmol·g-1·h-1·L-1,而有些为μmol·g-1·h-1,也造成结果无法直接对比。建议在未来研究中,反应均在室温和一个大气压下进行;对于粉末催化,产氨速率的单位用μmol·g-1·h-1或μg·g-1·h-1,对于电极催化,产氨速率的单位用μmol·g-1·cm-2或μg·g-1·cm-2。同时建议在实验方法中注明表观量子效率(AQE)测量的具体条件,包括光源强度、催化剂用量、反应体系体积等。
(2)发展高效的析氧助催化剂。理想的光催化氮还原的另一半反应为析氧反应,然而由于析氧反应四电子过程动力学缓慢,目前大多数LSP光催化氮还原是在有空穴牺牲剂的条件下进行的,而空穴牺牲剂的加入会给氨的测量带来干扰,同时也消耗了其它有机物分子。
(3)发展LSP金属/半导体界面精准调控技术。LSP金属/半导体界面结构直接决定着热电子的转移方式,通过界面原子和电子结构调控实现热电子的直接转移,提高热电转移效率。
(4)高活性和选择性氮气活化位点制备。目前LSP光催化氮气活化位点主要利用的是半导体的一些本征缺陷,例如氧化物中的氧空位和氮化碳中的氮空位,但是本征缺陷对氮气催化活性不够高,未来研究可以利用单原子制备技术在半导体上或LSP金属颗粒中引入对氮气具有高活性的单原子催化位点。
(5)LSP光热效应在光催化固氮中的研究。目前已经证明LSP局域光热效应可以提高氮气的转化率[64, 65],未来研究中可以通过精确调控LSP纳米颗粒和催化活性材料的空间排布,实现热量的有效管理[46],提高光催化固氮效率。
(6)LSP光催化氮气氧化研究。目前LSP光催化固氮都是基于氮气还原反应,理论上LSP光催化氮气氧化也是可行的,未来LSP光催化氮气氧化值得研究。
猜你喜欢
物理学报(2023年3期)2023-02-19
物理学报(2022年6期)2022-03-30
生态科学(2021年5期)2021-11-13
摄影之友(影像视觉)(2017年8期)2017-11-27
当代化工研究(2016年5期)2016-03-20
广西林业科学(2016年2期)2016-03-20
材料科学与工程学报(2016年5期)2016-02-27
——固氮微生物
新疆农垦科技(2016年3期)2016-02-20
天然气勘探与开发(2015年1期)2015-02-28
中国测试(2012年1期)2012-11-15
