
磁场中HD分子振转跃迁的超精细结构*
2021-09-17 06:08唐家栋刘乾昊程存峰2胡水明2
物理学报 2021年17期
唐家栋 刘乾昊 程存峰2) 胡水明2)†
1) (中国科学技术大学, 合肥微尺度物质科学国家研究中心, 合肥 230026)
2) (中国科学技术大学, 中国科学院量子信息与量子科技创新研究院, 合肥 230026)
HD分子红外跃迁的精密测量被用以检验量子电动力学、确定质子-电子质量比等.但HD分子的超精细结构分裂对于测量精度是一个很重要的限制因素, 并可能是实验中测得υ = 2—0谱带跃迁呈特殊线型的原因之一.本文分别在耦合表象和非耦合表象下计算了HD分子振转跃迁的超精细结构, 并计算了不同外加磁场下HD分子(2–0)带中R(0), P(1), R(1)线的超精细结构, 模拟了10 K低温下对应的光谱结构.结果表明, HD分子跃迁结构可随磁场发生明显变化.这可能有助于分析HD分子跃迁特异线型产生的机制, 进一步获得其准确的跃迁中心频率, 用于基础物理学检验.
1 引 言
分子的精密光谱对于大气探测[1]、检验量子电动力学(QED)[2−4]、测定基本物理常数[5]和探测物理常数的变化[6]等都有重要的意义.饱和吸收光谱是最常用的克服分子光谱多普勒展宽的实验方法[7−11].此时, 谱线的加宽主要来源于压力加宽、渡越时间加宽、功率加宽等.我们发展了基于腔增强和光频梳锁定的分子光谱测量技术, 对于分子近红外振转跃迁的饱和吸收可实现很高的测量精度[12].HD分子精密光谱的探测对于QED的检验、质子-电子质量比的确定有着重要的作用.我们之前用光梳锁定的腔衰荡光谱(CRDS)方法测量了HD第一泛频带R(1) (2–0)线的饱和吸收光谱,拟合得到的谱线中心位置[8]和Ubachs研究组[13]用噪声免疫腔增强光外差光谱(NICE-OHMS)方法测得的结果之间存在明显偏差.进一步的实验表明, HD分子跃迁饱和吸收谱呈明显的非对称线型[14,15].Ubachs研究组[14]认为, 这是HD分子饱和吸收中多个超精细分裂结构子能级间干涉叠加的结果.但该模型强烈依赖于分子间碰撞导致的布居转移, 这方面还缺乏有力的实验和理论支持.
60多年前, Quinn等[16]利用磁共振方法研究过HD分子基态的超精细结构, 近期, Dupre[17]和Komasa等[18]分别计算了HD分子红外跃迁的超精细结构, 在未考虑测量中泵浦探测效应的情况下, 得到了基本一致的跃迁结构, 但仍然难以预测实验条件下的光谱线型, 不能和实验结果进行直接对比.Pachalski等[19]认为, HD超精细结构的精密测量对于精确认识氘原子电四极矩, 理解自旋依赖的核子相互作用有着重要意义.本文提出, 通过在不同磁场下观测HD红外谱线结构的变化以检验其线型的模型.本文将计算不同磁场下HD分子υ = 2—0带中各塞曼子能级间电偶极跃迁的结构.由于分子间碰撞和能级布居转移的动力学效应对能级结构十分敏感, 通过实验和理论的比对, 有望判定HD分子红外跃迁呈现特别的非对称线型的真正机制.
2 计算方法
由于HD分子υ = 2—0谱带振转跃迁极其微弱(爱因斯坦系数A ≈ 10–5/s), 目前只有在腔增强光谱实验中才能得到其无多普勒的吸收光谱.这样的实验中, 比较方便的实验条件是加上一个和探测光同轴的外磁场.实验构想图如图1所示, 从ECDL激光器出来的探测光耦合进入一个光学共振腔, 腔外线圈产生一个与探测光同轴的磁场.利用光腔衰荡光谱[7]等腔增强光谱方法来测量HD分子在不同磁场下的饱和吸收光谱.
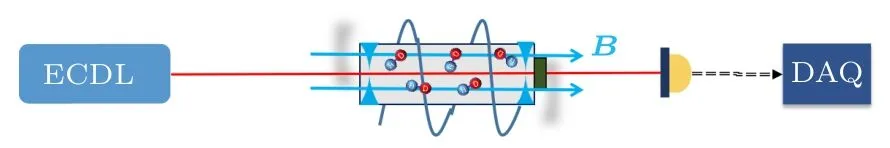
图1 在磁场中测定HD分子振转跃迁Fig.1.Determination of the ro-vibrational transition of HD molecule in magnetic field.
以下将分析HD振转跃迁在外磁场下的结构.为减小谱线的渡越加宽、增加下态布居, 比较理想的实验方式将是在低温下测量R(0)谱线.低温下HD分子主要布居在J = 0的转动能级, 相对来说,超精细结构较为简单, 也有利于分辨其光谱结构.因此, 本文将主要以υ = 2—0带中R(0)跃迁为例进行讨论.当然, 相关的计算方法很容易推广到其他跃迁.
可以用两种表象分别来计算HD分子振转跃迁的超精细结构.一种是选用N, iD, F1, iH, F,mF这6个量子数构成的耦合表象, 另一种是用N,mN, iH, mH, iD, mD这6个量子数构成的非耦合表象.其中N是转动角动量量子数, iD, iH分别是氘、氢原子核自旋角动量量子数, F1, F为耦合得到的量子数, mF, mN, mH, mD为对应的磁量子数.
2.1 耦合表象
耦合表象下, 选用N, iD, F1, iH, F, mF这6个量子数构成的基函数.此时, 可以认为HD分子的转动角动量N与氘原子核自旋角动量iD耦合为F1, F1再与氢原子核自旋角动量iH耦合为总角动量F, F在Z轴方向的投影记为mF, 如图2所示.即
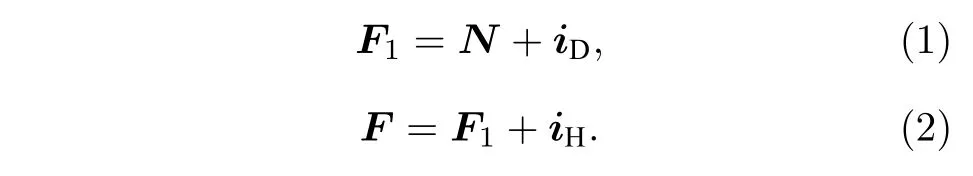
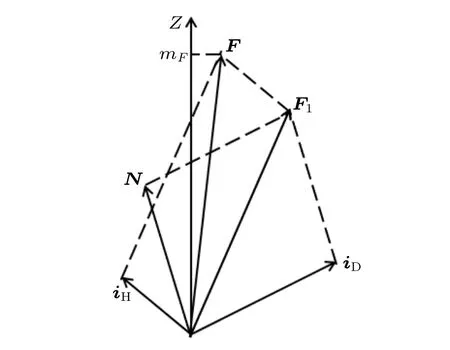
图2 耦合表象下HD分子的角动量耦合示意图Fig.2.Angular momentums of the HD molecule in the coupled representation.
在无外加磁场的情况下, 能级对于mF量子数是简并的.此时, 可以选择N, iD, F1, iH, F这5个量子数构成的表象来计算哈密顿矩阵.对于HD υ = 2—0带的R(0)跃迁, 上能级υ = 2, N = 1的态共有5组基.5组基│N iDF1iHF 〉 分别记为

系统的相互作用哈密顿量分为下面四个部分.第一部分为分子转动与氢原子核自旋的相互作用, 其中cH为氢原子核的自旋-转动常数:
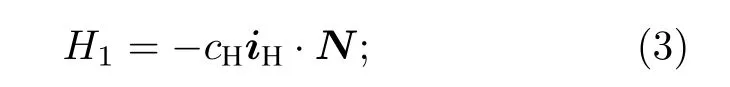
第二部分为分子转动与氘原子核自旋的相互作用,其中cD为氘原子核的自旋-转动常数:
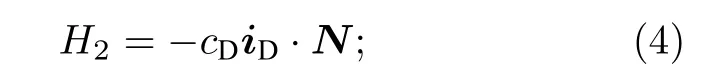
第三部分为由Breit方程[20]得到的两个原子核自旋磁矩之间的偶极-偶极相互作用, 其中cs为两个原子核自旋之间的相互作用常数, rHD为核间距,gH, gD分别为氢原子核和氘原子核的朗德因子,µN为核磁子, µ0为真空磁导率:
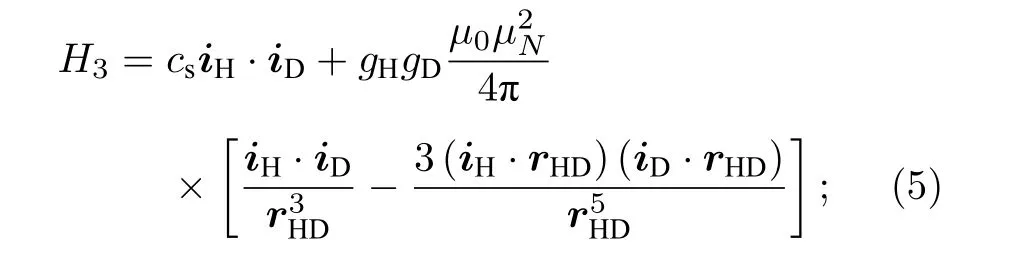
第四部分为电四极相互作用, Q为核电四极张量,∇E为静电场梯度:
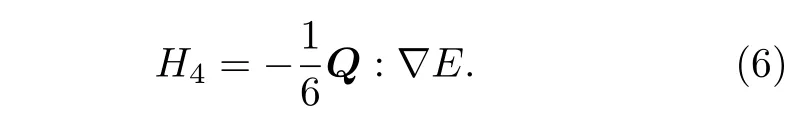
在N, iD, F1, iH, F这5个量子数构成的耦合表象下, 由于上能级υ = 2, N = 1只有5组基, 其哈密顿矩阵为一个5 × 5的矩阵.由于分子转动与核自旋之间的相互作用, 以及两个原子核自旋之间的相互作用, 哈密顿矩阵为一个非对角的矩阵.为了计算其矩阵元, 需要将系统的相互作用哈密顿量写成张量形式[21]:
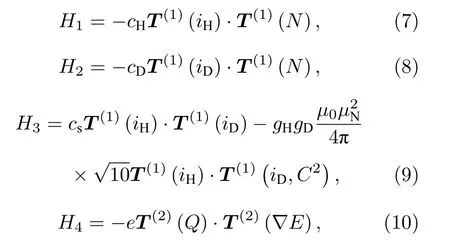
其中
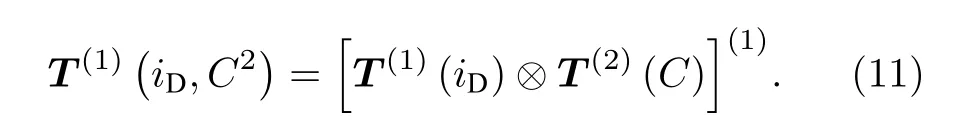
按照Dupre[17]的方法, 可以计算得到上能级的5 × 5的哈密顿矩阵中所有的矩阵元, 计算中用到的cH, cD等常数已经有了比较精确的计算结果[17,18,22].本文使用Komasa等[18]通过Born-Oppenheimer近似得到的结果, 上能级υ = 2, N =1对应的cH= 82.183 kHz, cD= 12.524 kHz, d1=16.654 kHz, d2= –22.043 kHz.得到的哈密顿矩阵的本征值即为各超精细分裂能级的能量移动, 本征向量即为对应的本征波函数.记5个本征态分别为:

对于下能级υ = 0, N = 0的态, 只有F = 1/2和F = 3/2两个能级, 且能量是简并的, 分别记为a态和b态.
各超精细跃迁谱线相对线强度主要由各跃迁电偶极矩的平方|µul|2决定, 计算下能级a, b两个态分别到上能级A, B, C, D, E五个态的跃迁电偶极矩的平方即可得到所有超精细跃迁谱线的相对线强度.
根据文献[17], 上下能级间的跃迁电偶极矩 ,µul=〈X||T(1)(µ)||Y〉=c1〈n||T(1)(µ)||Y〉+c2〈n′||T(1)(µ)||Y〉其中, X代表A, B, C, D, E中的一个态, Y代表a, b中的一个态, n和n' 代表1, 2, 3,4, 5中的一个态, c为归一化的叠加系数, 数值为对应的本征波函数的系数.〈 n||T(1)(µ)||Y〉 的值可以按照文献[17]中的公式计算得到:
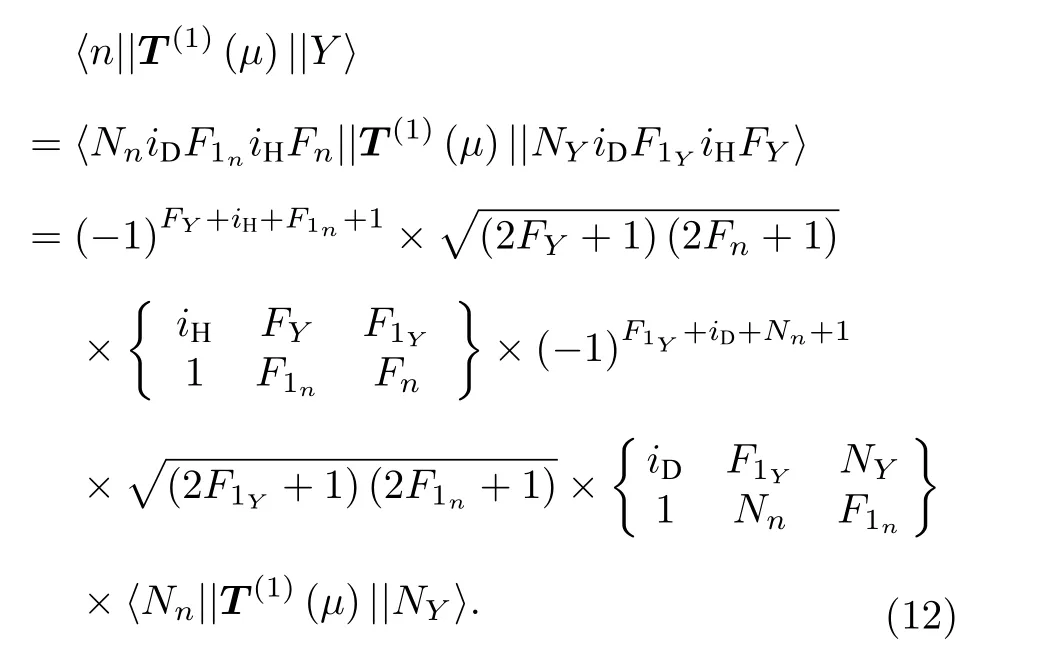
由于上能级Nn= 1, 下能级NY= 0, 最后一项 〈 Nn||T(1)(µ)||NY〉 的值对于所有超精细跃迁都是相等的, 只需计算前面的系数即可.例如, 要计算 b →A 的跃迁强度, 记其相对线强度为Sul, 由(12)式可以计算出, 〈 1||T(1)(µ)||b〉 和〈2||T(1)(µ)||b〉分别为由于 | A〉=−0.9493|1〉+0.3142|2〉 ,则
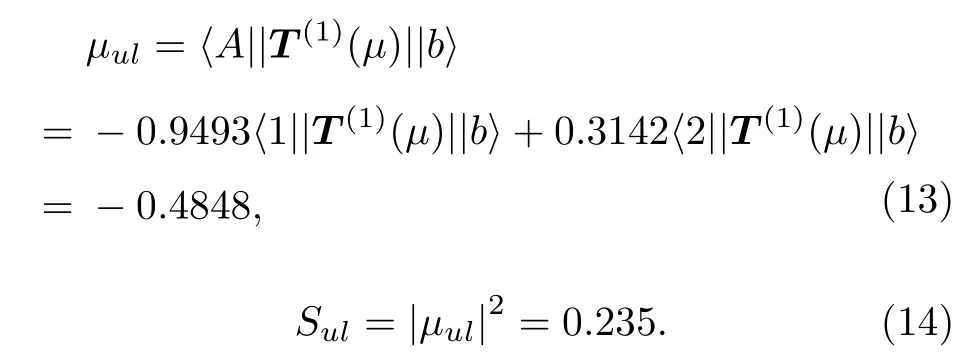
按照这种方法, 可以计算出HD分子υ = 2—0带R(0)线的所有超精细跃迁谱线的频率和相对线强度, 得到的结果如图3最下面一栏所示.如果进一步考虑Zeeman子能级间的跃迁, 其相对线强度为
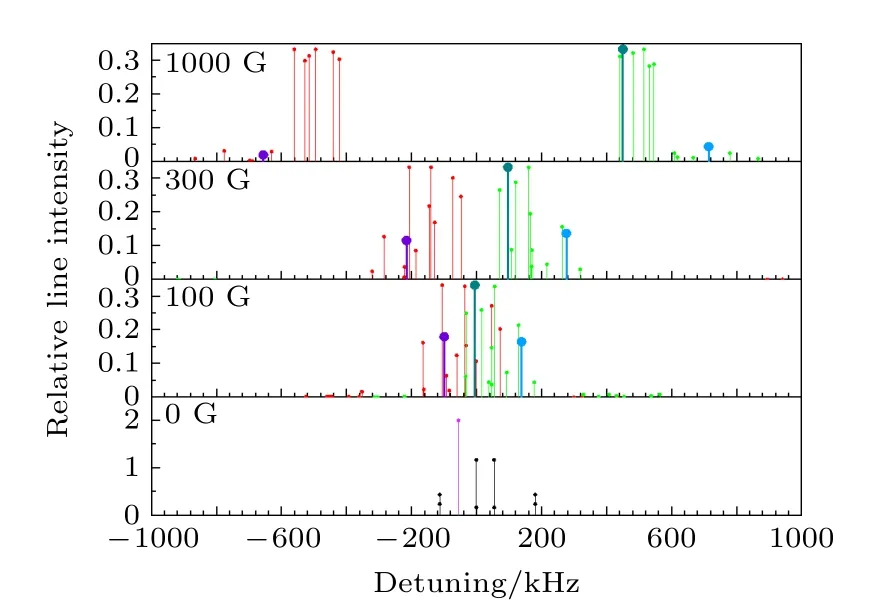
图3 计算得到的HD分子υ = 2—0带R(0)线的所有超精细跃迁谱线的频率偏移及其对应的相对线强度(有部分弱线在显示范围之外)Fig.3.Calculated frequency shifts of all hyperfine transition lines in the R(0) line in the υ = 2–0 band and their corresponding line intensities (some weak lines are outside the display range).
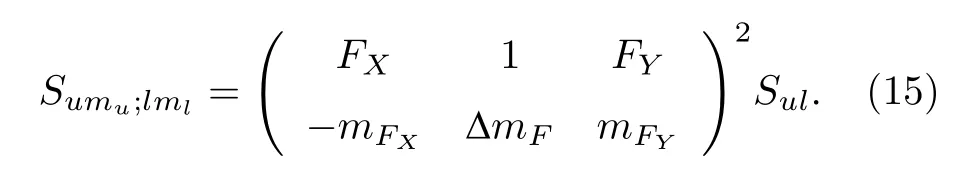
2.2 非耦合表象
也可以选择N, mN, iH, mH, iD, mD这6个量子数构成的非耦合基来进行计算.此时, 可以认为HD分子的转动角动量N、氘原子核自旋角动量iD以及氢原子核自旋角动量iH分别与磁场耦合.对于HD υ = 2—0带的R(0)线, 上能级υ = 2; N =1; mN= 0, ± 1; mH= ± 1/2; mD= 0, ± 1, 共有3 × 2 × 3=18组基.下能级υ = 0; N = 0; mN=0; mH= ± 1/2; mD= 0, ± 1, 共有2 × 3 = 6组基.在无外加磁场的情况下, 按照Ramsey和Lewis[22]的表示方法, 系统的有效哈密顿量为
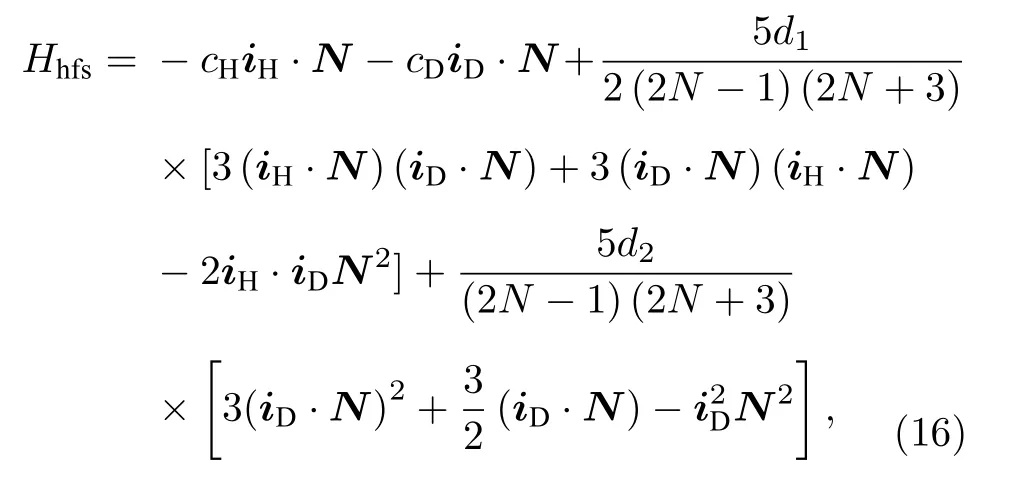
其中, 前两项对应于自旋-转动磁相互作用, 第三项对应于两个核自旋磁矩之间的偶极-偶极相互作用,第四项代表氘核电四极矩的相互作用.跃迁矩阵元可以按照角动量算符的性质来进行计算:
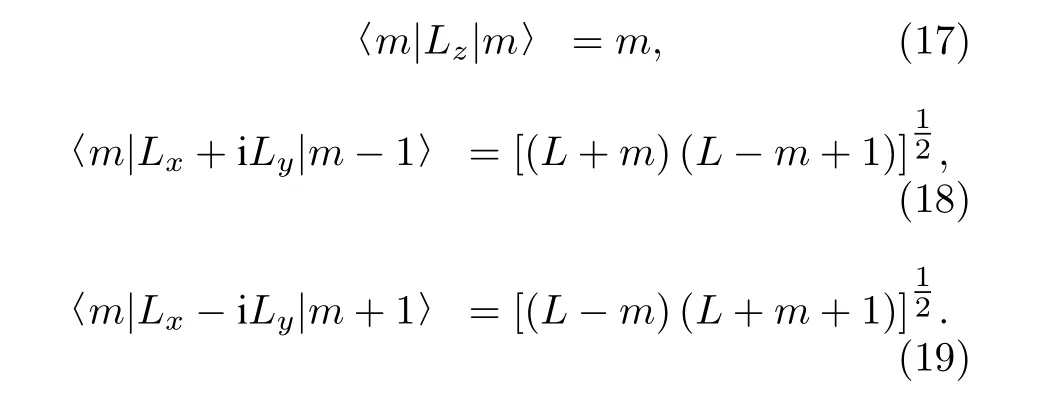
(16)式中角动量算符矢量积可以分别化为如下形式:
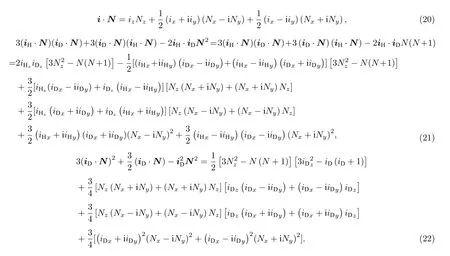
按照(17)式—(19)式给出的角动量算符的性质来计算有效哈密顿量Hhfs的矩阵元, 可以得到上能级18 × 18的哈密顿矩阵(注意是块对角化的).将得到的哈密顿矩阵求本征值和本征向量, 即可得到对应的各超精细分裂能级的能量移动和本征波函数, 非耦合表象基之间的跃迁矩可以由耦合表象基之间的跃迁矩得到.而对于下能级υ = 0,N = 0态的6个能级, 由于N = 0, 所以Hhfs= 0,能量是简并的, 且能量移动为0.
3 外磁场下的电偶极跃迁
在无磁场情形下, 可以分别用耦合表象和非耦合表象得到各超精细能级的位移和能级间的跃迁矩.我们计算得到的结果和文献[18]的结果一致.受限于光谱实验的分辨, 目前还没有HD振转跃迁的测量可以分辨超精细结构.例如, Hua等[15]测量了HD在υ = 2—0带R(1)线的光谱, 分辨率仅为800 kHz, 无磁场下完全无法分辨超精细结构.而有外加磁场时, HD超精细结构可以分辨开来,这为分辨超精细结构提供了契机.
在有磁场情况下, 采用非耦合表象更为方便.从N, mN, iH, mH, iD, mD这6个量子数构成的非耦合表象出发, 按照Ramsey和Lewis[22]的表示方法, 系统的有效哈密顿量为中H为磁场强度, 单位为其
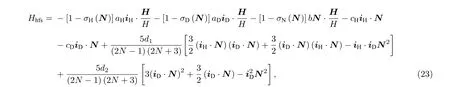
其中, 前两项对应于核磁矩与外加磁场的相互作用, 第三项为分子转动磁矩与外加磁场的相互作用, 第四项和第五项对应于自旋转动磁相互作用,第六项对应于两个核自旋磁矩之间的偶极-偶极相互作用, 第七项代表氘核电四极矩的相互作用.式他各系数的定义与文献[22]中的相同, 各系数的数值该文献中也有给出.
取磁场方向为Z方向, 由于光的方向与磁场方向相同, 这时只能探测Δm = + 1和Δm = –1的跃迁.因此以下只考虑这两种跃迁, 而不考虑Δm = 0的跃迁.Hhfs中前三项角动量算符只有Z方向的分量, 因此其对应的哈密顿矩阵元只有对角项不为零, 根据(17)式可以很容易算出.而后四项哈密顿矩阵元的计算与2.2节中的计算方法相同.得到哈密顿矩阵后, 求本征值和本征向量, 即可得到上、下能级分别在对应磁场下的各超精细分裂能级的能量移动和本征波函数, 由每条跃迁谱线上下能级的能量移动即可得到该跃迁谱线的频率移动.按照(13)式的叠加方法, 从非耦合表象基之间的跃迁矩按照叠加系数叠加即可得到对应超精细跃迁的跃迁矩, 进而得到每条超精细跃迁谱线的相对跃迁强度.在100, 300 和1000 G的外加磁场下计算得到的结果分别如图3上面三部分所示.结果的具体数值如表1所列.图3中频率偏移的零点对应于不考虑超精细分裂结构时的跃迁频率.相对跃迁强度的值取对应的跃迁电偶极矩的平方.在加上外加磁场后, 跃迁分为Δm = + 1和Δm = –1两支, 分别用红、绿两种颜色标注.为了显示某一条跃迁在考虑塞曼分裂后随磁场强度的变化, 选出了3条线分别用深紫、墨绿、蓝三种颜色的粗线标出, 这3条线都由无外加磁场中的紫色线分裂而出.
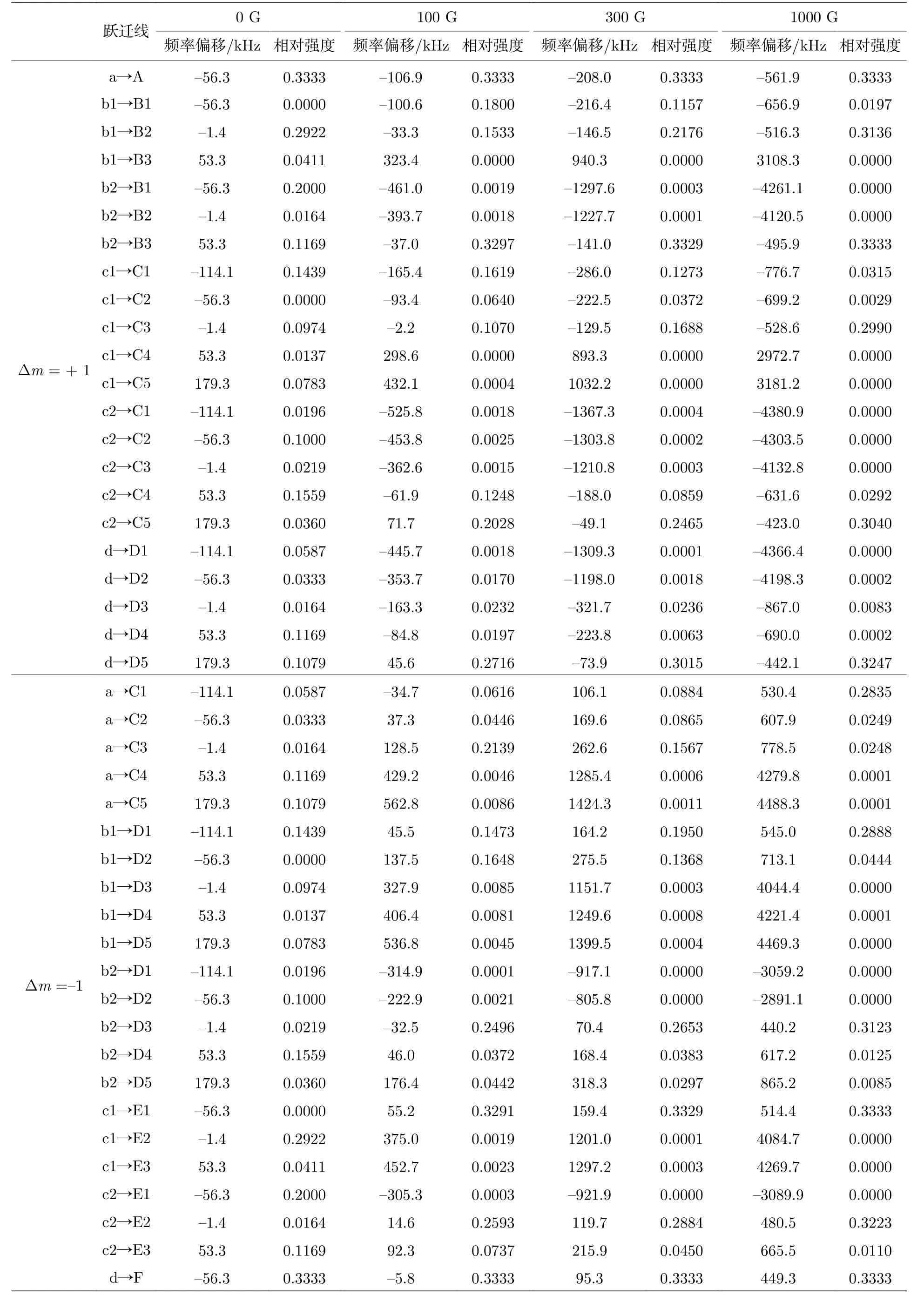
表1 计算得到的R(0)线所有超精细跃迁谱线的频率偏移及其对应的相对线强度Table 1.Calculated frequency shifts of all hyperfine transition lines in the R(0) line and their corresponding line intensities.
从图3可以看出, 当外加磁场强度较大时, 超精细跃迁谱线明显分为两支, 分别为Δm = + 1和Δm = –1的跃迁.随着外加磁场强度的增大,各超精细跃迁谱线频率偏移的数值也在增大.可以看出, 当外加磁场强度达到1000 G时, 两支超精细跃迁谱线已经完全分开, 此时分子转动角动量以及原子核自旋角动量之间的相互作用已经远小于外加磁场导致的磁能级的分裂, 此时使用非耦合表象更加直观.
同时, 我们也分别在无外加磁场以及100, 300,1000 G外加磁场的条件下, 按照上述的计算方法计算了HD分子υ = 2—0带中P(1), R(1)线的超精细分裂结构及各超精细跃迁谱线的相对线强度.得到的结果分别如图4和图5所示, 频率偏移的零点对应于不考虑超精细分裂结构时的跃迁频率, 相对跃迁强度的值取对应的跃迁电偶极矩的平方, 不同颜色的标注与图3中的意义相同.
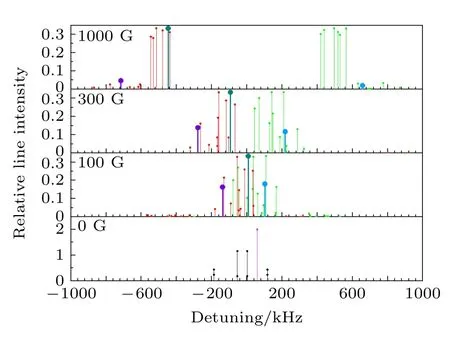
图4 计算得到的HD分子υ = 2—0带P(1)线的所有超精细跃迁谱线的频率偏移及其对应的相对线强度(有部分弱线在显示范围之外)Fig.4.Calculated frequency shifts of all hyperfine transition lines of υ = 2–0 band P (1) lines of HD molecule and their corresponding relative line intensities (some weak lines are outside the display range).
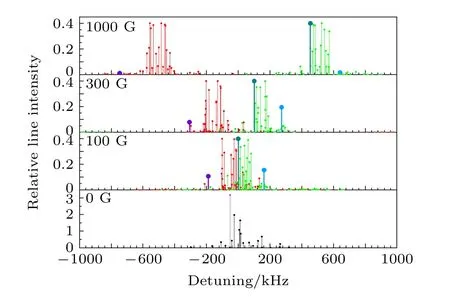
图5 计算得到的HD分子υ = 2—0带R(1)线的所有超精细跃迁谱线的频率偏移及其对应的相对线强度(有部分弱线在显示范围之外)Fig.5.Calculated frequency shifts of all hyperfine transition lines of HD molecule υ = 2–0 band R (1) line and their corresponding relative line intensities (some weak lines are outside the display range).
将计算得到的超精细跃迁谱线以相对强度为权重做加权平均, 可以发现两支超精细跃迁谱线叠加后的光谱中心位置的频率偏移与磁场强度都基本成线性关系, 斜率分别为0.5056和–0.5056 kHz/G,如图6所示.
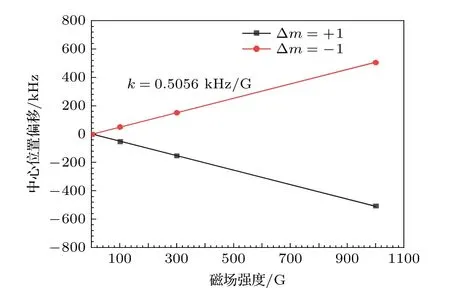
图6 HD分子R(0) (υ = 2—0)跃迁在轴向磁场下, Δm= + 1和Δm = –1两支超精细跃迁谱线光谱中心的频率偏移与磁场强度的关系Fig.6.Relationship between the magnetic field intensity and the frequency shift of the spectral center of the Δm =+ 1 and Δm = – 1 hyperfine transitions of the R(0) (υ =2–0) line of HD.
4 低温下光谱线型
在低压情况下, 每条超精细跃迁谱线的饱和吸收光谱的线宽可以近似认为是渡越时间加宽导致的.可以用公式[23]来估算渡越时间展宽的大小, 其中为分子的最概然速度.通过估算得到, 在10 K的温度下, 每条超精细跃迁谱线的半高全宽(FWHM)约为99 kHz.根据文献[24], 辐射场和吸收子之间的动量交换产生的recoil效应会导致每条超精细跃迁谱线的饱和吸收光谱分裂为两个峰.两个峰的中心位置分别位于未考虑recoil效应前该超精细跃迁谱线峰中心的两侧, 且偏移量相同, 记为对于HD分子υ = 2—0带的R(0)线, recoil效应产生的偏移量δ约为34 kHz, 即两峰间距约为68 kHz.
本文在理论上模拟了在10 K的低温条件下,分别在无磁场、100 G外加磁场、300 G外加磁场、1000 G外加磁场下HD分子υ = 2—0带R(0), P(1),R(1)线的光谱.模拟中只考虑了Δm = + 1和Δm = –1的跃迁.由于recoil效应产生的分裂, 每条超精细跃迁谱线都分为间距为2δ的两个峰.每个峰都用FWHM为99 kHz的洛伦兹线型来模拟,模拟得到的结果如图7所示.
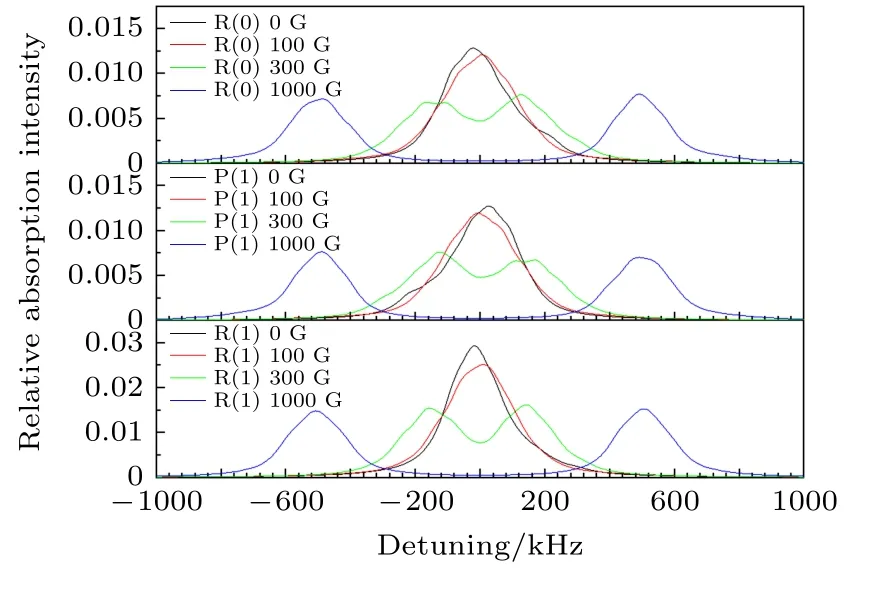
图7 在10 K的低温条件下, 分别在不同外加磁场下模拟的HD分子(2—0)带R(0)线、P(1)线、R(1)线的光谱Fig.7.Simulated spectra of R (0), P (1) and R (1) lines in the (2–0) band of HD under different magnetic fields at the temperature of 10 K.
5 结 论
为了检验超精细分裂结构对HD分子的光谱线型带来的影响, 在无外加磁场的条件下, 分别使用耦合表象和非耦合表象计算了HD分子(2—0)带中R(0), P(1), R(1)线的超精细分裂结构及各超精细跃迁谱线的相对线强度.利用无外加磁场得到的计算结果, 推导出了非耦合表象下各塞曼子能级之间的跃迁矩, 并分别在100, 300, 1000 G的外加磁场下, 计算出了R(0), P(1), R(1)线的所有超精细跃迁谱线的频率和相对线强度.模拟了在10 K的低温条件下, 在不同磁场下HD分子υ = 2—0带R(0), P(1), R(1)线的饱和吸收光谱.通过分析在低温下考虑渡越加宽和反冲分裂后叠加的光谱线型, 发现能级中心位置的频率偏移与磁场强度近似成线性关系, 斜率约为0.5 kHz/G.我们计划使用光梳锁定的腔衰荡光谱的方法, 在不同外加磁场的条件下, 测量常温下HD分子(2—0)带R(0), P(1),R(1)线的饱和吸收光谱, 与理论上模拟得到的结果进行对比, 来检验相关的光谱线型模型.
猜你喜欢
大学物理(2022年8期)2022-09-15
物理学报(2022年10期)2022-06-04
山西化工(2020年6期)2021-01-10
海洋通报(2020年2期)2020-09-04
复旦学报(医学版)(2020年3期)2020-06-18
原子与分子物理学报(2020年5期)2020-03-17
成长·读写月刊(2019年1期)2019-01-28
物理通报(2018年12期)2018-12-24
西华大学学报(自然科学版)(2018年2期)2018-04-04
中学生数理化·高三版(2017年1期)2017-04-20
