
Sr 掺杂羟基磷灰石负载Co3O4 催化N2O 分解
2021-09-10 07:09:00刘晓丽王永钊赵永祥
燃料化学学报 2021年8期
刘晓丽,王永钊,赵永祥
(1.山西大学精细化学品教育部工程研究中心,山西 太原 030006;2.山西大学化学化工学院,山西 太原 030006)
N2O 主要来源于硝酸和己二酸生产过程,化石燃料和生物质燃烧,以及汽车尾气的排放[1-3]。N2O 不仅加速了臭氧层的消耗,而且表现出强烈的温室效应,其全球变暖潜能值比CO2高310 倍[4]。随着经济社会的快速发展,N2O 排放量呈现出持续加速增长趋势[5]。因此,研究开发N2O 减排技术显得尤为重要。在众多消除N2O 方法中,直接催化分解因其效率高、不产生二次污染且成本相对较低等优势成为研究的重点[6]。
N2O 直接分解催化剂包括贵金属催化剂[7-9]、非贵金属氧化物催化剂[10-12]和离子交换沸石催化剂[13,14]。贵金属催化剂不仅催化效率高,且具有良好的稳定性,但高成本限制了其在N2O 催化分解中的应用[15]。离子交换沸石虽然拥有良好的催化活性,但当进料气中存在H2O 时,其结构不稳定导致催化活性降低[16]。非贵金属氧化物催化剂因其高活性、高稳定性、低成本等优势受到研究者的青睐[17],其中,最具代表性的尖晶石Co3O4含有两种不同价态的金属离子(Co3+、Co2+),可有效形成氧化还原对,表现出良好的催化性能。然而单一Co3O4催化剂在焙烧与反应过程中易烧结,从而造成催化活性下降[18]。掺杂Co3O4催化剂尽管由于Co3O4与掺杂氧化物间存在相互作用,表现出较高的催化活性和热稳定性,但N2O 分子的活化通常发生在气固界面,导致催化剂中尖晶石Co3O4活性相未被充分利用[19-21]。张涛等[22-24]制备了各种金属取代的六铝酸盐催化剂,深入研究了制备过程、晶体结构以及相组成等各种因素对催化剂催化N2O 分解性能的影响。发现在相同Mn 含量下,由于六铝酸钡中存在较高比例的八面体Mn3+,表现出比六铝酸镧更高的催化活性。同时,该课题组[25]还报道了将Fe3+掺杂到Ru 取代的六铝酸钡中,由于形成了稳定的BaRuO3相,从而抑制了Ru 的挥发,进一步提高了该催化剂的N2O 催化性能。众所周知,载体可以促进活性组分的分散、稳定与高效利用,同时载体适当的织构性质、酸碱性质对N2O 催化分解也具有重要影响[20-25]。Shen等[26]制备了各种氧化物载体负载四氧化三钴(Co3O4/AxOy,A=Mg、Zn、Mn、Al、Ce)催化剂,进一步研究了其N2O 催化分解活性。发现Co3O4/MgO 的N2O 催化活性最高,归因于Co 和MgO 之间的相互作用。Hu 等[27]研究了不同晶相ZrO2为载体的Co3O4/ZrO2催化N2O 分解性能。发现Co3O4/m-ZrO2明显优于Co3O4/t-ZrO2和单一Co3O4,这主要是由于较高的比表面积和增强的Co2+/Co3+氧化还原性能。可见,载体可以改变催化剂的织构性质,促进活性组分的分散和热稳定性,从而增强其催化性能。
化学计量羟基磷灰石Ca10(PO4)6(OH)2(HAP),由于其稳定性高、酸碱性可调、成本低、易于离子交换且可回收性好而被广泛用作催化剂材料[28]。HAP 中的Ca2+可被Zn2+、Mg2+、Sr2+、La2+、Cd2+、Pb2+、Cu2+和Fe2+等离子所取代,进而调变HAP 的热稳定性、相稳定性、比表面积以及表面反应性等[29]。近年来,离子掺杂磷灰石被广泛用于制备各种反应的催化剂。Shanthi 等[30]利用水热法合成了具有棒状形貌的Sr 掺杂羟基磷灰石(SrHAP-R),并通过浸渍法制备了Ni/SrHAP-R。由于Sr 掺杂增强了催化剂的碱性,在山梨糖醇催化分解制乙二醇和1,2-丙二醇的反应中,乙二醇收率高达60%。Ogo 等[31]在水热条件下制备了具有不同Sr/P 物质的量比的锶羟基磷灰石(Sr-HAP),并用于催化乙醇合成1-丁醇。研究发现,具有高Sr/P 物质的量比的Sr-HAP 显示出更高的催化活性和1-丁醇选择性。这可归因于高Sr/P 物质的量比催化剂较高的碱性位点密度。Christopher 等[32]对比研究了不同碱土金属掺杂羟基磷灰石上丙酮合成甲基异丁基酮反应,发现与Ba2+和Mg2+相比,离子半径适中的Sr 掺杂羟基磷灰石表现出更高的催化性能。基于上述报道可见,Sr 掺杂羟基磷灰石不仅本身是一种优良催化剂,同时也是一种性能良好的催化剂载体,但是将其用于负载型Co 基催化剂催化N2O 分解的研究尚未见报道。
在这项工作中,采用简单的共沉淀法来制备一系列Sr 掺杂羟基磷灰石,通过浸渍法制得负载型Co3O4/Sr-HAP 催化剂,并研究了其deN2O 分解性能。同时,利用XRD、N2-physisorption、Raman、H2-TPR、XPS、O2-TPD 和CO2-TPD 等手段对所制备样品进行表征,详细研究了Sr/Ca 原子比对Co3O4/Sr-HAP 催化剂结构、织构、活性位和表面性质的影响。
1 实验部分
1.1 实验原料
Co(NO3)2·6H2O,分析纯,天津市光复科技发展有限公司;Ca(NO3)2·4H2O,分析纯,天津市光复科技发展有限公司;(NH4)2HPO4,分析纯,北京化工厂;NH3·H2O,分析纯,国药集团化学试剂有限公司;Sr(NO3)2,分析纯,天津市恒兴化学试剂制造有限公司。
1.2 载体和催化剂的制备
HAP 和Sr-HAP 的制备:以(Ca+Sr):P=1.67 计算Sr(NO3)2、Ca(NO3)2·4H2O 和(NH4)2HPO4用量。将一定量的Sr(NO3)2、Ca(NO3)2·4H2O 溶解在250 mL去离子水中并加热至60 ℃,以获得均匀的溶液,其中,Sr/Ca 原子比分别为0、1∶9、2∶8 和3∶7。在连续搅拌下,将溶解在250 mL 去离子水中的(NH4)2HPO4溶液加入到上述溶液中。进一步通过缓慢滴加浓氨水将混合溶液的pH 值调至11。将所得悬浮液继续搅拌2 h,然后老化24 h。过滤悬浮液,同时用去离子水充分洗涤,直至滤液pH 值达到7。将获得的滤饼在空气中于120 ℃干燥过夜,随后在空气气氛下600 ℃焙烧2 h,制得Ca10(PO4)6(OH)2、Ca9Sr1(PO4)6(OH)2、Ca8Sr2(PO4)6(OH)2和Ca7Sr3(PO4)6(OH)2,分别标记为HAP、Ca9Sr1、Ca8Sr2和Ca7Sr3。
催化剂的制备:首先,将3.899 g Co(NO3)2·6H2O分别溶解在7.5、6.0、6.0 或9.0 mL H2O 中;然后,将15 g HAP、Ca9Sr1、Ca8Sr2或Ca7Sr3加入到上述溶液中并充分搅拌,将获得的混合物在室温下老化3 h;最后,将样品在120 ℃下干燥过夜,并在空气气氛下600 ℃焙烧2 h。所得催化剂标记为Co/HAP、Co/Ca9Sr1、Co/Ca8Sr2和Co/ Ca7Sr3(催化剂的理论Co 含量为5%)。
1.3 催化剂的表征
样品的X 射线衍射谱图用Bruker D8 Advance X-ray 衍射仪测定。放射源CuKα 射线(λ=0.154 nm),10°-80°扫描,扫描速率2.4(°)/min。
样品的氮气物理吸附采用Micromeritics ASAP-2020 型物理吸附仪测定。将载体和催化剂样品于150 ℃真空脱气5 h,然后在-196 ℃进行氮气吸附。并分别采用BET 程序和BJH 方法测定、分析所测样品的比表面积(ABET)、平均孔径和孔体积。
样品的拉曼光谱使用Horiba Scientific LabRam HR Erolution 拉曼光谱仪获得。激发波长为532 nm,输出功率为10 mW。光谱为100-800 cm-1,分辨率为1 cm-1。
样品的FT-IR 光谱采用Bruker Tensor 27 傅里叶变换红外光谱仪获得。测试之前,将所有样品各取1 mg,然后用100 mg 真空干燥的IR 级KBr稀释。最后以4 cm-1的分辨率在400-2000 cm-1对样品进行16 次扫描。
样品的透射电镜(TEM)图像采用JEM-2100透射电子显微镜测定,加速电压200 kV。
样品的X 射线光电子能谱(XPS)采用ESCALAB 250 光谱仪分析测试,以位于283.1 eV 的C 1s信号作为基准,对XPS 数据进行处理,以校正结合能。
H2-TPR 在Micromeritics AutoChemII 2920 分析仪上进行。样品用量为30 mg (40-60 目),在5%H2/N2(30 mL/min)混合气氛下以10 ℃/min 程序升温至700 ℃,并记录H2-TPR 谱图。
O2-TPD 在Micromeritics AutoChemII 2920 化学吸附分析仪进行测定。将300 mg 样品在反应管中300 ℃用30 mL/min 的O2预处理1 h,然后在O2气氛中降温至50 ℃。将气体切换为He (30 mL/min)吹扫,待基线平稳后,在He 气氛下以5 ℃/min 程序升温至550 ℃,并记录O2-TPD 谱图。
CO2-TPD 测试使用的仪器与O2-TPD 相同。将100 mg 样品在He 气氛中(30 mL/min)加热至300 ℃并恒温1 h。然后将样品冷却至50 ℃并进行CO2脉冲直至吸附饱和。在He 气氛(30 mL/min)中将温度升至500 ℃(升温速率10 ℃ min-1),同时记录CO2-TPD 谱图。
1.4 催化剂的活性评价
N2O 催化分解利用连续流动微反应器于常压下测试。催化剂用量300 mg (40-60 目,体积约为0.3 mL),将流量为50 mL/min 的0.1% N2O/Ar 混合气体通过催化剂,空速(GHSV)10000 h-1。利用Agilent 7890B 气相色谱仪分析反应器入口和出口处的N2O 含量。N2O 转化率的计算公式如下:
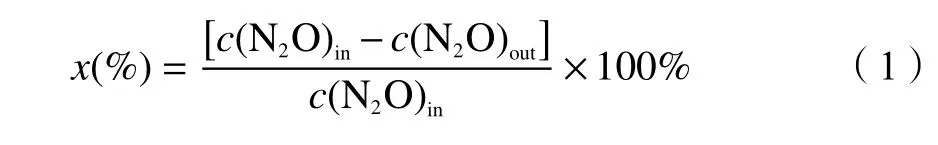
式中,x为N2O 的转化率,c(N2O)in为反应器入口的N2O 含量,c(N2O)out为反应器出口的N2O含量。
根据以下公式计算催化剂的TOF 值:

式中,p0为大气压(101.325 kPa),F(mL/s)为原料气体混合物的总流量,R为气体常数(8.314 J/(mol·K)),mcat为样品的质量,T0即室温(298 K),n(μmol/g)是通过O2-TPD 计算得到的活性位点的量。
2 结果与讨论
2.1 催化剂的XRD 表征
载体和催化剂的XRD 谱图分别如图1(a)和(b)所示。从图1(a)可看出,纯HAP 载体在10.8°、25.9°、31.8°、32.2°、32.9°、34.0°、39.8°、46.6°、48.1°、49.5°和53.2°处出现了明显的衍射峰,归属为HAP 的特征衍射峰(PDF#74-0565)。与纯HAP的XRD 谱图对比可发现,Ca9Sr1和Ca8Sr2的衍射峰峰型无明显变化,但位置略微向低角度方向移动,表明在保持了Ca10(PO4)6(OH)2结构的情况下,Sr 已成功掺入HAP 结构。随Sr 掺杂量进一步增加,Ca7Sr3的衍射峰主要位于25.9°、31.0°、31.8°和34.2°,且峰形较为弥散,与纯HAP 差别较大,表明Ca7Sr3以无定形存在。可见,当Sr/Ca 比达到3∶7时,载体中HAP 结构已遭到破坏。
图1(b)中显示,Co/HAP、Co/Ca9Sr1和Co/Ca8Sr2的XRD 谱图均出现了除HAP 特征峰以外的其他衍射峰。在2θ=36.8°处的衍射峰,归属于尖晶石结构Co3O4相的(311)晶面(PDF#09-0418)[33]。与Ca7Sr3样品XRD 谱图相比,除归属Co3O4衍射峰外,Co/Ca7Sr3样品中位于17°和30°的衍射峰归属于Ca2P2O7·4H2O 相。同时,随着Sr 掺杂量的逐渐增加,Co3O4衍射峰强度逐渐变弱,说明其晶粒尺寸变小,分散度提高。但与Ca7Sr3相比,Co/Ca7Sr3的XRD 谱图中载体衍射峰明显增强。推测这是由于Sr 掺杂量较高时Ca7Sr3以无定形存在,在催化剂制备过程中,经进一步焙烧导致其结晶度增加所致。与此同时,利用Scherrer 方程计算了四种催化剂中Co3O4的平均晶粒尺寸,并列于表1 中。
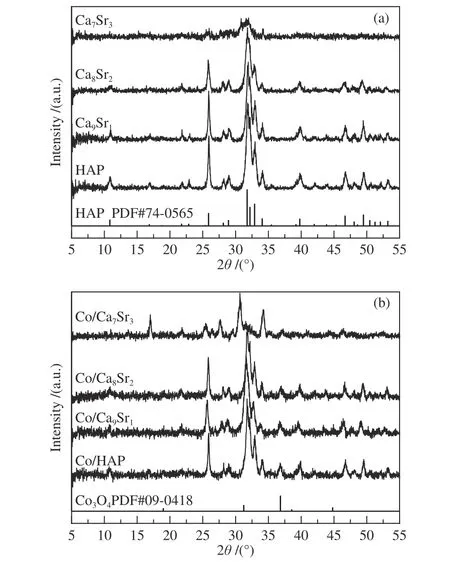
图1 载体(a)和催化剂(b)的XRD 谱图Figure 1 XRD patterns of supports (a) and catalysts (b)

表1 载体和催化剂的织构性质以及Co3O4 的晶体尺寸Table 1 Textural properties of the supports and catalysts as well as crystal size of Co3O4
2.2 催化剂N2 adsorption-desorption isotherms 表征
图2(a) 和(b) 分别给出了载体和相应催化剂的N2吸附-脱附曲线和孔径分布。根据IU-PAC分类,Ca7Sr3和Co/Ca7Sr3属于III 型等温线。除此之外,其他样品在相对压力p/p0高于0.8 时,氮气吸附量均急剧增加,且具有H3 迟滞回线,属于IV 型等温线,表现出典型的介孔结构[34],归因于颗粒聚集所形成的粒间孔[35]。在孔径分布(图2(b)插图)中,除Ca7Sr3和Co/Ca7Sr3外,其他载体和催化剂的孔径分布均处于10-100 nm。表1 汇总了不同样品的比表面积、孔容和平均孔径。可以看出,随Sr 掺杂量的增加,载体的比表面积逐渐减小,分别为44、33、27 和7 m2/g。与HAP、Ca9Sr1和Ca8Sr2相比,Ca7Sr3比表面积急剧减小。结合XRD表征结果,推测这是由于较多Sr 的掺杂破坏了HAP 结构所致。同时,与载体相比,相应催化剂的比表面积和孔体积均有所减小,这可归因于载体部分孔道被活性组分覆盖或堵塞[36]。
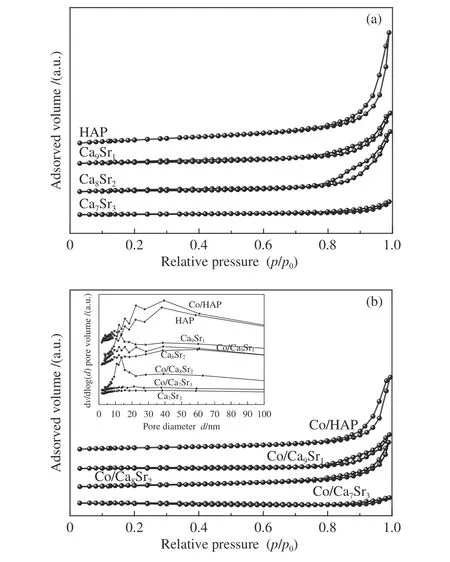
图2 载体(a)和催化剂(b)的N2 物理吸附-脱附曲线和孔径分布Figure 2 N2-physisorption isotherms and pore size distribution of supports (a) and catalysts (b)
2.3 催化剂的Raman 表征
图3 给出了催化剂的拉曼光谱谱图。图中位于962 cm-1附近的拉曼峰归属于载体羟基磷灰石结构中P-O 的V1 反对称伸缩振动模式[37]。与Co/HAP相比,Co/Ca9Sr1、Co/Ca8Sr2和Co/Ca7Sr3在962 cm-1处的拉曼峰宽化,且位置略微向低波数区域位移,同时Co/Ca8Sr2的强度明显降低,表明掺杂Sr 后载体结晶度降低,这与XRD 表征结果一致。此外,图中所有样品的Raman 谱均在195、477、517、615和685 cm-1处出现了五个Raman 峰,分别对应于Co3O4的3×F2g、Eg和A1g振动模式[38]。通过比较各催化剂的拉曼光谱可以看出,除Co/Ca7Sr3外,Co/HAP、Co/Ca9Sr1和Co/Ca8Sr2三个催化剂上Co3O4的拉曼峰宽度依次增加且峰强度降低,表明Co3O4纳米颗粒的分散性提高,这同样与XRD表征结果相一致。

图3 催化剂的拉曼光谱谱图Figure 3 Raman spectra of catalysts
2.4 催化剂的FT-IR 表征
图4 为载体和相应催化剂的FT-IR 谱图。载体HAP、Ca9Sr1和Ca8Sr2在400-1000 cm-1均显示出典型的磷酸盐吸收带,分别归属为P-O 反对称伸缩振动模式V1(965 cm-1),对称伸缩振动模式V3(1097、1045 cm-1)以及O-P-O 弯曲振动模式V4(601、565 cm-1)和V2(471 cm-1)[39]。然 而,载体Ca7Sr3在该区域的典型吸收带均向低波数位移,表明载体中Ca10(PO4)6(OH)2结构已遭到破坏。同时,各载体在3436-3446 和1633 cm-1处的谱带归因于表面吸附分子水的伸缩和弯曲振动模式[39]。此外,3575 和630 cm-1处的峰均归属于羟磷灰石骨架中OH 的振动,随Sr 掺杂量增加,3575 cm-1处OH 振动峰强度逐渐减弱,在Ca7Sr3红外谱中基本消失,进一步表明该载体中Ca10(PO4)6(OH)2结构遭到破坏。通常情况下,尖晶石Co3O4中归属为Co3+-O 和Co2+-O伸缩振动的两个FT-IR 特征谱带分别在665 和580 cm-1处出现。值得注意的是,各催化剂与其相应载体的红外光谱相比未发生明显变化,但在664 cm-1处出现对应于Co3+-O 伸缩振动的谱带,而未观察到Co2+-O 的代表性谱带,这是由于被载体的O-P-O(601 cm-1)弯曲振动谱带覆盖所致。

图4 载体和催化剂的红外光谱谱图Figure 4 FT-IR spectra of supports and catalysts
2.5 催化剂的TEM 表征
载体和相应催化剂的TEM 表征如图5 所示。可以看出,纯HAP 载体的形貌为长棒状。当Sr/Ca比为1∶9 时,与HAP 相比,Ca9Sr1的形貌基本保持不变,仍为长棒结构。随Sr 掺杂量进一步增加,Ca8Sr2变为短棒状,而Ca7Sr3棒状结构消失。从TEM照片估算了载体的粒径尺寸,HAP、Ca9Sr1、Ca8Sr2和Ca7Sr3的长度分别大致集中在78-86、75-84、8-15 和13-22 nm。四个催化剂样品的TEM 照片显示,Co/Ca8Sr2催化剂保持了载体的基本形貌,并出现了一些呈分散和聚集状态的Co3O4颗粒。对于Co/Ca9Sr1和Co/HAP 催化剂,聚集的Co3O4颗粒增加。当Sr/Ca 比为3∶7 时,载体原来的形貌完全消失,并且观察到了明显聚集的Co3O4颗粒。可见,与其他三种催化剂相比,Ca8Sr2上Co3O4颗粒分散程度相对较高,这可能是由于Co3O4与Ca8Sr2之间存在较强的相互作用[40]。

图5 载体和催化剂的TEM 照片Figure 5 TEM images of supports and catalysts
2.6 催化剂的XPS 表征
催化剂的XPS 谱图如图6 所示。对于Co/HAP,位于782.4、779.5 和781.1 eV 处的Co 2p3/2峰分别归属于为α(Co2+)、β(Co3+)和ε(离子交换Co2+)。在约788.6 eV 处检测到卫星峰(γ),表明存在Co2+[41]。
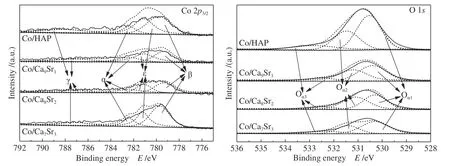
图6 催化剂的XPS 谱图Figure 6 XPS spectra of catalysts
与Co/HAP 相比,随Sr 掺杂量的增加,Co/Ca9Sr1和Co/Ca8Sr2的β 峰轻微向低结合能方向移动,表明Co3+周围的电子云密度降低,从而削弱了Co3+-O键[42]。但随着掺杂量的进一步增加,Co/Ca7Sr3的β 峰明显向高结合能方向移动。通过分峰拟合Co 2p3/2的峰面积计算出Co2+/Co3+之比列于表2。Co/HAP表面Co2+/Co3+比为1.25,随Sr 掺杂量的增加,Co/Ca9Sr1和Co/Ca8Sr2的Co2+/Co3+比进一步增大,分别为1.40 和1.69。然 而,当Sr/Ca 比 为3∶7 时,Co/Ca7Sr3的Co2+/Co3+比值急剧降至0.96。可见,与其他三个催化剂相比,Co/Ca8Sr2表面Co2+含量更高。

表2 催化剂的XPS 表征Table 2 XPS results of catalysts
四种催化剂的O 1s谱图如图所示,位于530.3-530.4、531.1-531.4 和532.3-532.9 eV 的O 1s峰分别归属为表面晶格氧(Oα1),吸附在氧空位上的表面氧物种(Oα2)和OH-或H2O 中的氧物种(Oα3)。表2 中包含了根据O 1s光谱拟合峰面积计算的所有样品的Oα2/(Oα1+Oα2+Oα3)值。从表2 中可以看出,Co/HAP 催化剂中的Oα2/(Oα1+Oα2+Oα3) 比 为0.31,随Sr 掺杂量的增加,Co/Ca9Sr1和Co/Ca8Sr2的该值进一步增大,分别为0.34 和0.45。然而当Sr 掺杂量进一步增加时,Co/Ca7Sr3中此值则显著降低。可见Co/Ca8Sr2中表面氧空位数量相对更多。
2.7 催化剂的H2-TPR 表征
图7 给出了载体和相应催化剂的H2-TPR 谱图。由图7 可知,各载体在100-750 ℃均未出现还原峰。对于催化剂而言,在100-700 ℃条件下H2-TPR曲线相似,均出现三个峰。280-380(α1)和380-550 ℃(α2)的还原峰分别归属于Co3O4中Co3+到Co2+的还原和Co2+到Co0的还原。同时,各催化剂在高于 600 ℃出现了还原峰β,归因于取代了羟基磷灰石结构中Ca2+的Co2+的还原[28],这是在浸渍过程中通过离子交换形成的Co 物种。

图7 载体和催化剂的H2-TPR 谱图Figure 7 H2-TPR profiles of supports and catalysts
表3 给出了各催化剂还原峰的位置和相应的H2消耗量。由表3 可知,与其他催化剂相比,Co/Ca8Sr2的α1还原峰明显向低温移动,表明Co3+更容易还原为Co2+,Co-O 键削弱;同时,还原峰α2峰顶温度升高至450 ℃,这意味着Co2+更难被还原为Co0。从氧化还原的角度来看,Co/Ca8Sr2中的Co2+表现出较弱的接收电子能力,也就是说,其供电子能力增强[40]。四种催化剂中β 还原峰H2消耗量明显不同,Co/HAP、Co/Ca9Sr1和Co/Ca7Sr3的H2消耗量分别为98.1、99.0 和9.1 μmol/g,而Co/Ca8Sr2的β 峰的H2消耗量最多,高达106.2 μmol/g。可见Co/Ca8Sr2含有更多通过离子交换形成的Co2+。众所周知,羟基磷灰石中易于交换的Ca 处于八面体配位环境,即由5 个O 和1 个OH 组成的近似八面体。因此,与Ca 交换的Co2+处于同样的配位环境[41]。与尖晶石结构的Co3O4中的Co2+相比,离子交换形成的Co2+更易于失去电子,因而难以还原[42]。Wang等[43]分别用无活性或低活性的Zn2+、Al3+和Fe3+取代Co3O4中Co2+和Co3+,研究了不同Co 离子在苯氧化中的催化活性,发现Co2+Oh(八面体Co2+)比Co2+Td(四面体Co2+)更容易被氧化成Co3+物种,同样表明与四面体Co2+相比,八面体Co2+更易于失去电子。
2.8 催化剂的O2-TPD 表征
图8 给出了四种催化剂的O2-TPD 谱图。由图8 可知,各样品在所测温度范围内均出现两个脱附峰。其中,位于50-300 ℃的脱附峰是由吸附在表面氧空位上氧的脱附形成;另一个在300-550 ℃的峰则归属于表面晶格氧的脱附[44]。Co/HAP 上表面氧物种从91.6 ℃开始脱附,峰顶温度出现在140.6 ℃。与Co/HAP 相比,Sr 掺杂催化剂上表面氧物种起始脱附温度差别不大,但随Sr 掺杂量增加,其峰顶温度向低温方向位移,表明Sr 掺杂样品上氧物种迁移率增加,脱附更容易。

图8 催化剂的O2-TPD 谱图Figure 8 O2-TPD profiles of catalysts
就脱附峰面积而言,随Sr/Ca 比增加,催化剂的O2脱附峰面积逐渐增大,但当Sr/Ca 为3:7 时,O2脱附峰面积则明显减小。基于脱附峰面积计算了各催化剂表面氧物种的脱附量。Co/HAP、Co/Ca9Sr1、Co/Ca8Sr2和Co/Ca7Sr3上脱附量分别为40.9、84.3、101.6 和20.7 μmol/g。可见,Co/Ca8Sr2催化剂表面具有更多的吸附氧物种,表明存在更多的氧空位,这与XPS 结果一致。
2.9 催化剂的CO2-TPD 表征
图9 为催化剂的CO2-TPD 谱图。从图9 可看出,所有曲线在50-550 ℃均出现两个脱附峰。通常情况下,在50-300 和300-550 ℃的脱附峰分别归因于吸附在弱碱性位点以及强碱性位点上二氧化碳的脱附[42]。注意到除Co/Ca7Sr3催化剂外,随Sr 掺杂量增加,催化剂上CO2脱附峰面积逐渐增大,表示碱量增多。根据脱附峰面积计算了各催化剂上两种碱性位的数量,结果如表4 所示。

图9 催化剂的CO2-TPD 谱图Figure 9 CO2-TPD profiles of catalysts
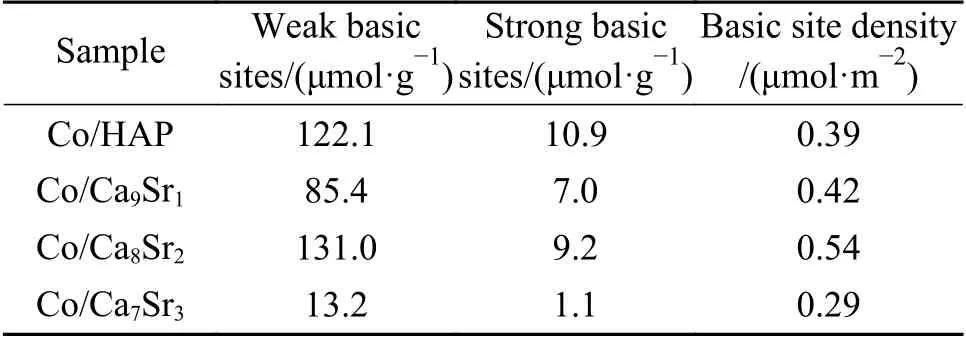
表4 催化剂表面上不同碱性位点的数量和碱性位点密度Table 4 Amount of different basic sites and basic site density on the catalyst surface
与Co/HAP 相比,随Sr 掺杂量的增加,催化剂上碱性位数量先增加后减少,其中,Co/Ca8Sr2催化剂上的碱性位数量最多,高达131.0 μmol/g。进一步结合比表面积计算了各催化剂上碱性位点密度。结果显示,四种催化剂上碱性位点密度按如下顺序递减,Co/Ca8Sr2> Co/Ca9Sr1> Co/HAP >Co/Ca7Sr3。可见,与其他催化剂相比,Co/Ca8Sr 催化剂具有相对更高的碱性位密度。
2.10 催化剂的反应性能评价
载体和相应催化剂的N2O 催化分解活性结果见图10。由图10 可知,各载体在280-600 ℃均未表现出催化活性。与其他催化剂相比,Co/Ca8Sr2表现出最优的N2O 分解活性。Co/Ca8Sr2上N2O 分解开始于约280 ℃,并在520 ℃实现完全转化。Co/Ca9Sr1和Co/HAP 上N2O 完全转化温度分别提高至560 和580 ℃,而Co/Ca7Sr3的催化性能明显降低,在反应温度高达600 ℃时N2O 仍未完全转化。此外,通过O2脱附量计算了Co/HAP、Co/Ca9Sr1、Co/Ca8Sr2和Co/Ca7Sr3催化剂在340 ℃时的TOF值,分别为5.18×10-5、6.12×10-5、7.67×10-5和5.08×10-5s-1。可见,HAP 载体中适量Sr 的掺杂可明显提高Co/Sr-HAP 催化剂的N2O 催化分解性能。

图10 载体和催化剂的N2O 催化分解性能Figure 10 N2O conversion over the supports and catalysts feed gas:1× 10-3 N2O/Ar,GHSV=10000 h-1
通常,尖晶石氧化物Co3O4上N2O 的分解属于氧化还原反应过程[45]。其分解机理为,首先Co2+提供电子给N2O 分子反键轨道,削弱N-O 键,并使其断裂进而形成N2和Co3+O-(式(1)),然后通过L-H 机理,两个Co3+O-扩散并结合成O2分子,O2脱附后再得到Co2+(式(2)),从而完成催化循环。此外,Co3+O-也可以直接与N2O 反应,通过ER 机理形成Co2+、N2和O2(式(3))。可见,尖晶石氧化物Co3O4的N2O 催化分解性能与Co2+的给电子能力,O2的脱附能力以及催化剂中Co2+的含量等密切相关[42]。


XRD 结果表明,掺入少量Sr 后,Ca9Sr1和Ca8Sr2仍然保持了HAP 的晶体结构,而Ca7Sr3由于Sr 掺杂量过多致使HAP 结构遭到破坏,这同样可由FT-IR、TEM 等表征所证实。同时,XRD、Raman与TEM 等表征结果显示,与其他催化剂相比,Co/Ca8Sr2中Co3O4具有更高的分散度和较小的晶粒尺寸。N2物理吸附结果表明,催化剂的比表面积随着Sr 掺杂量增加,先降低又增加,然后又降低,孔容逐渐减小,这与其催化活性变化不一致,表明比表面积并不是影响催化剂在该分解反应中的主要因素。进一步XPS、TPR 和O2-TPD 等表征则证实四种催化剂表面均含有Co3+、Co2+以及离子交换的Co2+,其中,除Co/Ca7Sr3外,随Sr 掺杂量的增加,催化剂中Co2+和表面氧空位数量均增加,且Co/Ca8Sr2中离子交换的Co2+数量最多,氧物种更易于脱附。此外,Co/Ca8Sr2不仅拥有更多的碱量,而且具有更高的碱性位密度,这同样有利于增强Co2+的给电子能力。结合Co 基催化剂上N2O 分解反应机理,可推测除分散度、晶粒尺寸、比表面积等因素外,Co/Ca8Sr2上更多的离子交换形成的Co2+,更易于脱附的氧物种,以及高的碱性位密度等是其表现出相对优异N2O 分解活性的关键因素。
3 结 论
研究了Sr 掺杂羟基磷灰石对负载Co3O4催化剂载体和活性组分晶相结构、织构、形貌、表面物种和碱性质的影响,同时测试了催化剂N2O 催化分解活性。少量Sr 的掺杂在保持HAP 载体结构的基础上,促进了Co3O4分散,增加了Co2+尤其是离子交换Co2+和表面氧空位的数量,且氧物种更易于脱附,同时催化剂的表面碱量及碱性位密度也显著提高,这对于N2O 催化分解具有重要作用。因此,Co/Ca8Sr2表现出更优的N2O 催化分解活性。
猜你喜欢
湿法冶金(2019年5期)2019-10-18 09:00:00
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:45
浙江农业科学(2016年11期)2016-05-04 04:16:45
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
电源技术(2015年2期)2015-08-22 11:27:56
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
湖北科技学院学报(医学版)(2015年3期)2015-02-28 19:43:51
深圳大学学报(理工版)(2015年5期)2015-02-28 16:21:34
无机化学学报(2014年4期)2014-02-28 17:31:23
