
基于生物信息学筛选和鉴定结直肠癌 关键的生物标志物
2021-09-09 01:07:20张永玲范文涛
激光生物学报 2021年4期
张永玲,范文涛
(陕西中医药大学基础医学院,咸阳 712046)
结直肠癌是临床上最常见的消化道恶性肿瘤之一,致死率在恶性肿瘤中居第三位。根据2018年全美的癌症统计数据显示:男性和女性的结直肠癌发病率在各类肿瘤发病率中均居于第三位[1]。该疾病多发生在中年以上的男性,以40~70岁最为常见。随着人口老龄化以及生活习惯等因素的影响,结直肠癌的发病率和死亡率呈逐步上升趋势,且越发年轻化,但其发病原因仍然不明。现代临床早期诊断主要检测癌胚抗原(carcinoembryonic antigen,CEA)或糖类抗原199(carbohydrate antigen199,CA199)、糖类抗原242(carbohydrate antigen242,CA242)、糖类抗原724(carbohydrate antigen724,CA724),其虽可以诊断结直肠癌,但敏感度不高。越来越多的研究数据发现,基因异常和突变的基因参与结直肠癌的致癌作用和进展,如p53基因、原癌基因K-ras突变、表皮生长因子受体(epidermal growth factor receptor,EGFR)、Toll样受体(toll-like receptor,TLR)、同源异型盒D10(homeoboxD10,HOXD10)基因等。p53是一种重要的肿瘤抑制基因,通过调节不同的下游基因,在多种信号的转导过程中发挥着重要的作用[2]。原癌基因K-ras是表皮生长因子受体信号通路的下游分子,是引发和治疗结直肠癌的关键基因[3]。有相关研究发现,有近50%的结直肠癌患者其K-ras基因均发生突变[4]。TLR4是跨膜传递信号的受体,可以识别病原病参与相关的免疫应答[5]。根据部分临床研究发现,TLR4位于外显子区域的1196C>T及896A>G两个位点的突变最为明显,其可以改变氨基酸已被编码的序列[6]。也有研究表明,TLR4基因的多态性对炎性因子的合成过程产生影响,进而引发结直肠癌[7]。郗昌磊等[8]运用蛋白免疫印迹法(Western blot,WB)检测结直肠癌细胞中HOXD10的表达,发现过表达的HOXD10可以明显的降低Notch信号通路Notch1及下游靶基因Hesde的蛋白的表达,由此得知过表达的HOXD10可能通过下调Notch信号通路进而影响结直肠癌的发生。目前已发现的可用于诊疗的分子靶点很多,但仍需不断寻找有效的靶点。结直肠癌的早期症状不明显,晚期其发病率和死亡率在消化系统的恶性肿瘤中仅次于胃癌、食管癌和原发性肝癌。因此,开发有效的诊断和治疗方法至关重要。在近几十年里,微阵列技术和生物信息学分析日益广泛用于各类疾病相关基因的分析层面,也更好地帮助我们识别差异表达基因和功能通路参与结直肠癌的致癌作用和进展。然而,通过分析独立的微阵列数据很难获得可靠的结果。因此,在本研究中,从基因表达(gene expression omnibus,GEO,http://www.ncbi.nlm.nih.gov/geo)数据库下载3个数据集,并通过分析获得结直肠癌组织和非肿瘤组织之间的差异基因;然后,通过京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)通路富集分析和蛋白质相互作用(protein-protein interaction,PPI)网络分析进一步帮助我们理解致癌分子机制。综上方法所述,472个差异基因和15个核心基因通过分析被确定为有可能是结直肠癌候选的生物标志物。
1 材料和方法
1.1 GEO数据库
GEO是一个公共功能基因组学高通量基因表达数据的数据存储库,包括芯片和微阵列数据[9]。我们从GEO数据库筛选了3个基因表达数据集GSE21510、GSE32323、GSE15781(均来自于GPL570平台)。GSE21510数据集包含104个结直肠癌组织样本和44个癌旁样本;GSE32323包含22个结直肠癌样本和22个非肿瘤样本;GSE15781包含25个结直肠癌样本和17个非肿瘤样本。
1.2 筛选差异基因
选择GEO数据库自带分析工具Analyze with GEO2R进行分组,分为癌组织组和正常组织组,点击TOP250进行分析,保存分析数据。根据P<0.01,|logFC|>1筛选出差异基因。
1.3 绘制韦恩图
采用韦恩图制作软件(venny2.1),将检索出的差异基因绘制韦恩图,查找3组共有基因。
1.4 对共有差异基因进行KEGG和GO富集功能分析
DAVID (http://david.ncifcrf.gov)[10]是一个在线的可视化的生物信息数据库,整合了生物数据和分析工具,为用户提供更加全面的基因和蛋白质功能注释信息,可从中提取生物学信息。KEGG数据库是系统分析基因产物在细胞中的代谢途径以及这些基因产物功能的数据库[11]。基因本体论(gene ontology,GO)是一个主要的生物信息学工具,可以注释基因和分析基因的生物过程[12]。运用DAVID在线数据库可以将差异基因的功能和生物分析结果更好地以可视化的方式呈现。P<0.05被认为具有统计学意义。
1.5 PPI网络蛋白互作分析(筛选核心基因)
PPI网络是使用搜索工具(http://string-db.org)来检索基因相互作用的在线数据库[13]。通过分析功能蛋白之间的相互作用可以为疾病产生的机制或疾病的发展提供新的见解。STRING数据库是一个常用的 PPI数据库,combine score>0.4被认为是具有统计学意义。Cytoscape是可视化的分子相互作用网络,为用户提供了一个开放的生物信息学软件平台,内含的插件MCODE(molecular complex detection)是一个应用程序集群,其通过基于给定的差异基因蛋白互作网络图发现蛋白互作密集区域,即核心基因所在区域[14-15]。选择的标准如下:MCODE score>5,degree cut-off=2,node score cut-off=0.2,max depth=100,k-score=2。核心基因的KEGG和GO分析模块使用DAVID可视化呈现。
1.6 核心基因选择和功能分析
运用Cytoscape中的插件MCODE选择核心基因。网络的基因分析使用cbioportal(http://www.cbioportal.org)在线平台[16]。使用Kaplan-Meier曲线(cbioPortal)对核心基因的总体生存和无病生存进行分析。
1.7 核心基因验证
使用数据库Oncomine(http://www.oncomine.com)将正常组织与癌症组织进行对比分析。
2 结果与分析
2.1 筛选差异基因
从GEO数据库中筛选3个与结肠癌相关的数据集,通过对数据集中的数据统一处理之后,鉴定出3个数据集的差异表达基因:GSE21510(148个样本)、GSE32323(44个样本)、GSE15781(42个样本)。其中GSE21510中有4 993个差异表达基因,GSE32323中有2 504个差异表达基因,GSE15781中有1 096个差异表达基因。通过韦恩图(venny2.1)可见3个数据集包含了472个重复基因,其中包括上调基因212个,下调基因260个(图1a)。
2.2 对差异基因进行KEGG和GO富集功能分析
运用DAVID在线可视化数据库分析差异基因的功能和富集结果。利用GO分析,将所有差异基因富集到生物学过程(biological process,BP)、细胞组分(cellular component,CC)和生物学功能(molecular functions,MF)3种生物关系中。分析结果表明,蛋白在生物学过程中主要参与调节细胞增殖、免疫应答、离子传输、调节细胞活性、脂质的分解过程等(表1)。细胞学组分主要集中细胞外区域等。生物学功能主要集中于蛋白结合、肽酶的活性、细胞骨架的绑定以及结构分子活动。KEGG通路分析结果显示,下调的差异基因主要富集在过氧化物酶体增殖物激活受体(peroxisome proliferator activated-receptor,PPAR)信号通路、细胞因子与细胞因子受体的相互作用以及醛固酮调节钠的重吸收,而上调的差异基因主要富集于淀粉和蔗糖代谢TGF-β信号通路等。

表1 差异基因KEGG和GO富集功能分析Tab. 1 KEGG and GO enrichment function analysis of differential gene
2.3 PPI网络蛋白互作分析(筛选核心基因)
使用STRING数据库、Cytoscape在线分析软件对差异基因和核心基因的PPI网络进行构建(图1b、1c)。运用Cytoscape中的插件MCODE,根据score排序,选择出得分最明显的区域图1 c,即核心基因。通过DAVID对核心基因的功能进行分析。GO分析结果显示,核心基因在生物学过程中主要参与蛋白耦合受体蛋白信号通路和细胞表面受体信号转导,生物学功能主要集中于趋化因子受体结合、细胞和生长因子活动(表2)。KEGG主要富集在趋化因子信号通路以及受体的互动。

表2 核心基因KEGG和GO富集功能分析Tab. 2 Enrichment function analysis of core genes KEGG and GO
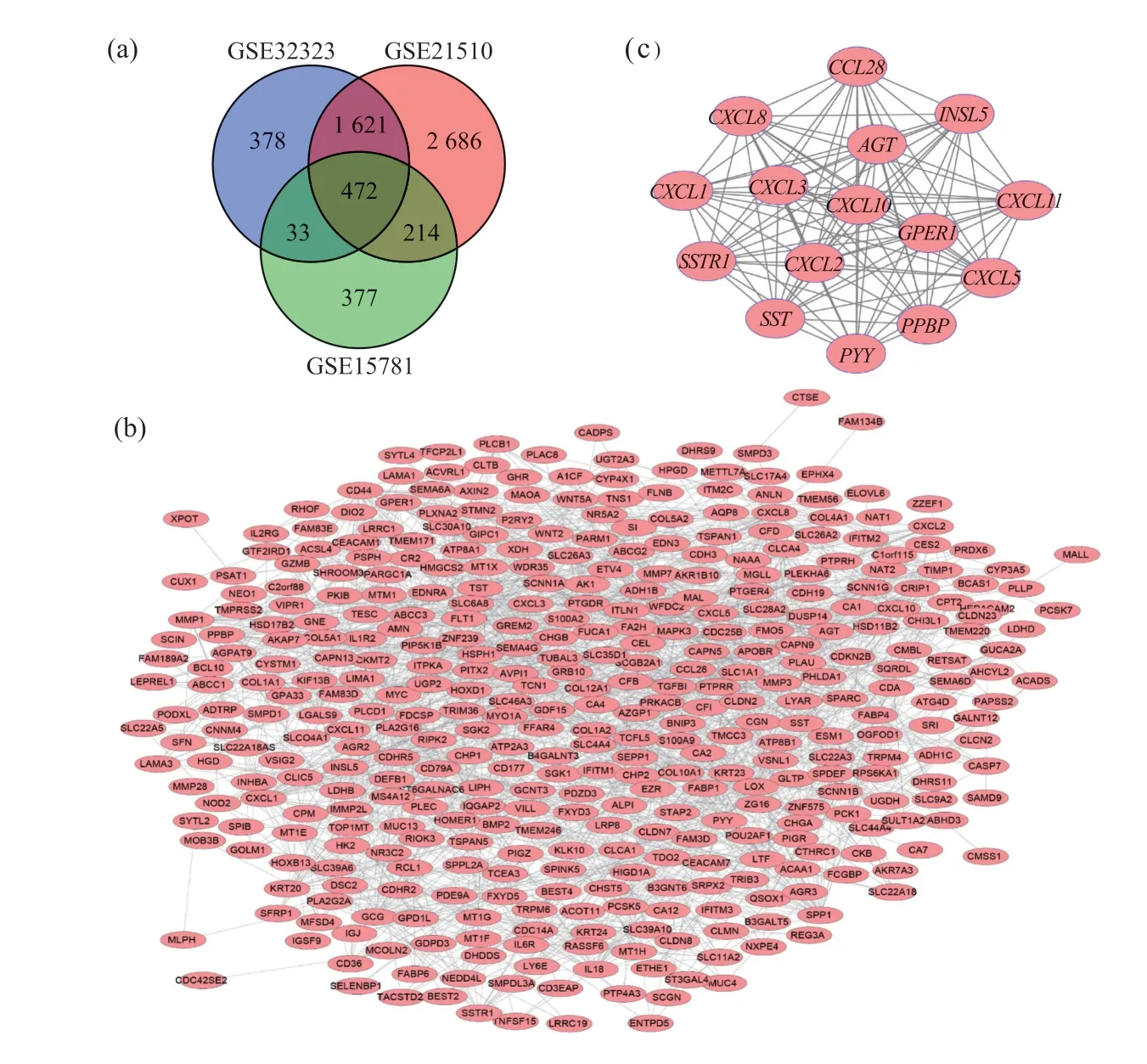
图1 差异基因的韦恩图、PPI网络和核心基因Fig. 1 Venn diagram, PPI network and core gene diagram of differential genes(a)在mRNA表达谱集GSE21510、GSE32323和GSE15781中选择倍数变化>2且P<0.01的基因。3个数据集显示472个基因的重叠,即472个差异基因;(b)使用Cytoscape构建差异基因的PPI网络;(c)从获得的PPI网络中筛选出最重要的模块,即核心基因。 (a) Select genes with fold change>2 and P value<0.01 in the mRNA expression profile sets GSE21510, GSE32323 and GSE15781. The 3 data sets show an overlap of 472 genes, that is, 472 differential genes; (b) Use Cytoscape to construct a PPI network of differential genes; (c) Screen out the most important module, the core gene, from the obtained PPI network.
2.4 核心基因选择和功能分析
总共15个基因被确定为核心基因。这些核心基因的名称、缩写和功能如表3所示。使用cbioPortal在线平台对核心的基因构建共表达基因蛋白网络(图2a)。使用Kaplan- Meier曲线对核心基因的总体生存进行分析(图2 b),发现结直肠癌患者含有CXCL2、AGT基因,其生存率相比无病生存率较差。

图2 核心基因的相互作用网络和生存曲线分析Fig. 2 Analysis of interaction network and survival curve of core genes(a)使用cbioPortal分析核心基因及其共表达基因。黑色粗体轮廓的节点代表核心基因,黑色细体轮廓的节点代表共表达基因;(b)使用cbioPortal在线平台对核心基因进行总体存活和无病存活分析。 (a) Use cbioPortal to analyze Hub genes and their co-expressed genes. The nodes with black bold outlines represent core genes, the nodes with the black body outline represent co-expressed genes; (b) Use the cbioPortal online platform to analyze the overall survival and disease-free survival of core genes.
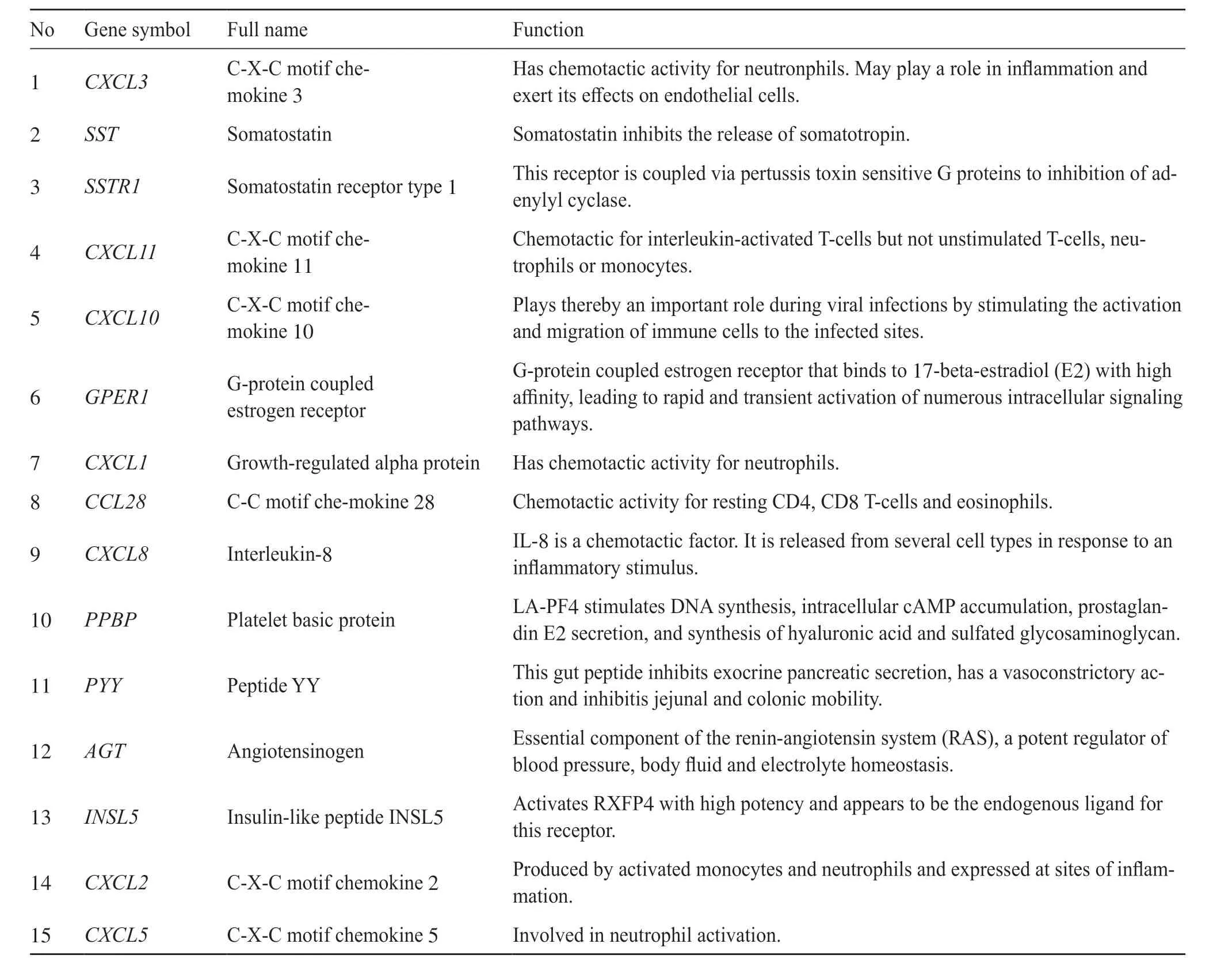
表3 核心基因功能Tab. 3 Core gene functions
2.5 核心基因的验证
利用数据库Oncomine将基因AGT与CXCL2在正常结直肠组织中的表达与结直肠癌组织中的表达进行对比,结果用箱式图显示(图3)。选取Oncomine数据库中3个数据集进行进一步验证(图4),结果发现,AGT与CXCL2在结直肠癌组织确实呈高表达。
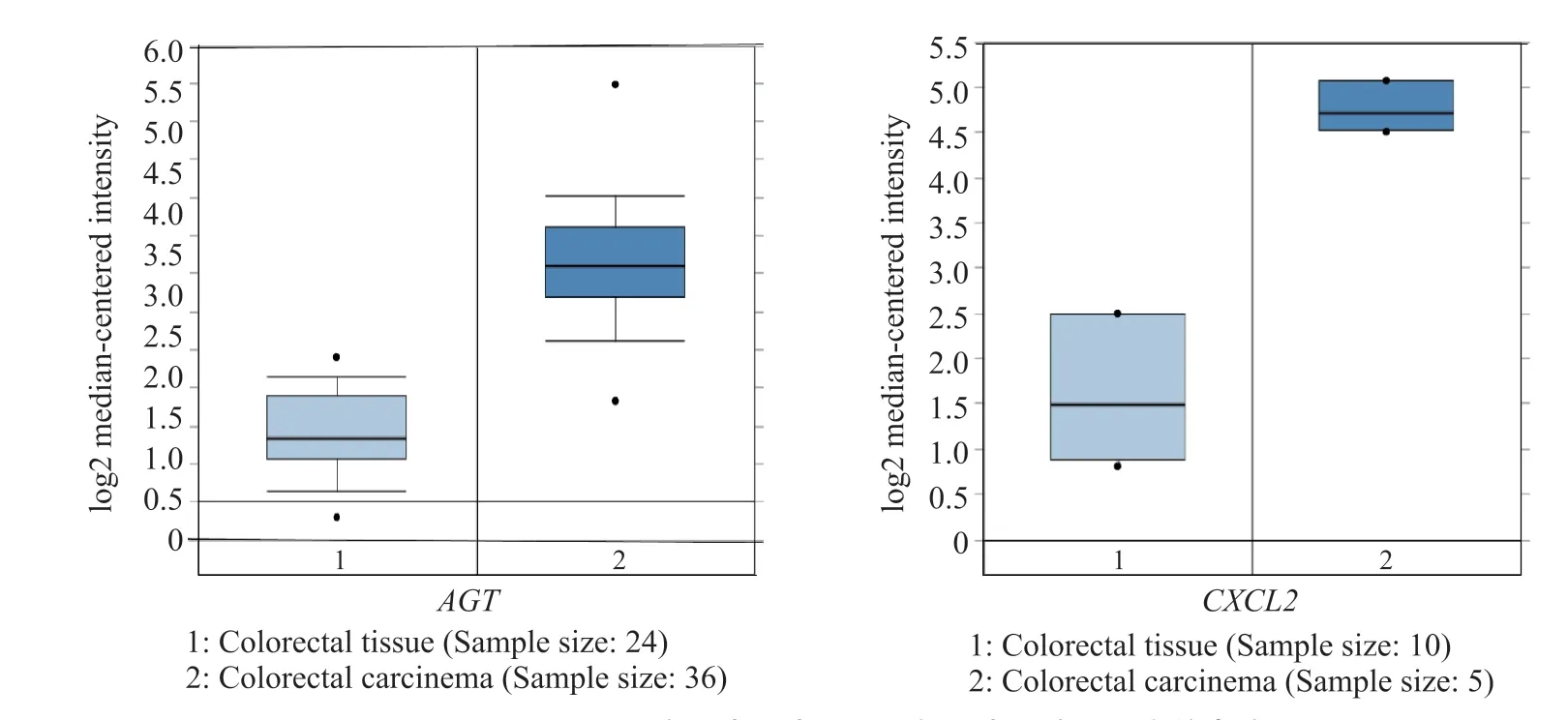
图3 AGT和CXCL2在正常结直肠组织与结直肠癌组织中的表达Fig. 3 The expression of AGT and CXCL2 in normal colorectal tissues and colorectal cancer tissue
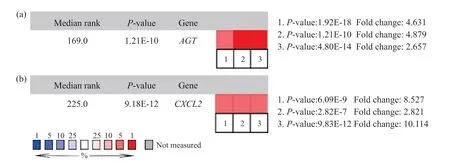
图4 AGT和CXCL2在Oncomine 3个数据集中的验证结果Fig. 4 The verification results of AGT and CXCL2 in Oncomine’s three data sets(a)AGT;(b)CXCL2。1.大肠癌与正常Graudens结肠的比较;2.直肠癌与正常Skrzypczak结直肠的比较;3. 结肠癌与正常Skrzypczak结肠直肠比较。Oncomine数据集结果显示AGT、CXCL2在结直肠癌组织中呈现明显的高表达。(a) AGT; (b) CXCL2. 1. Colorectal carcinoma vs. normal graudens colon; 2. Colorectal carcinoma vs. normal skrzypczak colorectal; 3. Colon carcinoma vs. normal skrzypczak colorectal. The results of the Oncomine dataset showed that AGT and CXCL2 were significantly over-expression in colorectal cancer tissues.
3 讨论
结直肠癌已经成为中国三大癌症之一,绝大多数的结直肠癌来源于腺瘤,其发病率正以螺旋速度递增,但其病因仍然模糊不清。结直肠癌并非不可防治,是最易自我筛查的疾病,但其潜伏期较长,早期诊断率较小。
近几年有关结直肠癌分子机制的研究还是甚少。本研究基于GEO数据库,选择3个数据集,筛选出差异基因和核心基因,并对其蛋白互作网络、分子功能以及生存曲线通过cytoscape、cbioportal等生物信息工具进行分析,最终根据分析结果确定了AGT、CXCL2基因对结直肠癌具有促进作用。就目前,在我们所能查阅到的文献中,只有1篇文献研究了AGT与结直肠癌的相关性:AGT不仅可以调节血压和稳定体液平衡,而且能够抑制肿瘤细胞的增殖、迁移;AGT M235T多态性与中国人结直肠癌的分化程度密切相关[17]。本研究结果与上述研究结果有所出入,分析原因如下:1)样本量有限,所以有必要扩大样本量;2)我们在GEO数据库中所选取的数据集中的样本主要来自于日本和挪威,不能排除这种差异是由种族的差异而引起的,所以需要在扩大样本量的基础上增加多种族的样本来源,来获得更加准确的分析结果;3)我们的研究是一个描述性的研究,还有待功能验证。CXCL2是一种趋化因子,与CXCR2受体结合参与细胞的增殖和凋亡、肿瘤的形成及其发展,以及干细胞的分化、中性粒细胞的聚集、神经递质的释放。宋宏伟等[18]运用免疫组化染色分析结直肠癌及癌旁组织中CXCL2蛋白的表达,结果发现CXCL2表达可能通过某种特殊的途径影响结直肠组织,从而致癌,但其具体途径还有待研究。郑敏[19]运用免疫印迹法、酶联免疫吸附法(enzyme linked immunosorbent assay,ELISA)检测发现,结直肠癌组织及血清中的CXCL2蛋白表达均明显高于癌旁正常组织。当根据解剖部位分为结肠、直肠时,CXCL2在结肠的高表达比在直肠更加明显[20]。CXCL2联合检测CEA、CA199能够有效的诊断结肠癌[21]。
本研究旨在识别可能参与结直肠癌的致癌或进展的差异基因。通过对472个差异基因和15个核心基因的识别,发现促癌因子AGT、CXCL2可能被视为对结直肠癌诊断的生物标志物,为后续的研究提供分子靶标。然而,未来还需要进一步的研究来阐明这些基因在结直肠癌细胞中的生物功能及其激活途径。
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
心电与循环(2020年1期)2020-02-27 07:48:24
中国医药指南(2017年3期)2017-11-13 02:56:23
哈尔滨医药(2016年3期)2016-12-01 03:58:33
癌变·畸变·突变(2016年3期)2016-02-27 06:15:25
西南军医(2016年3期)2016-01-23 02:17:57
中国继续医学教育(2015年6期)2016-01-07 07:38:47
中国当代医药(2015年30期)2015-03-01 02:08:01
湖北农业科学(2014年3期)2014-07-21 10:25:00
西南军医(2014年1期)2014-02-03 03:06:31
